Zusammenfassung: Mit dem Vektorimpfstoff Ad26.COV2.S (Johnson & Johnson; Janssen Biotech Inc.) erhielt am 11.3.2021 der 4. Impfstoff gegen COVID-19 eine bedingte Marktzulassung durch die Europäische Kommission (EC). Dieser Entscheidung liegt die Zwischenanalyse einer noch laufenden randomisierten kontrollierten Studie zugrunde, nach der die klinische Wirksamkeit – Verhinderung mittelschwerer und schwerer Verläufe von COVID-19 – bei 67% liegt. Reaktogenität und frühe Nebenwirkungen sind ähnlich wie beim ersten zugelassenen Vektorimpfstoff ChAdOx1 nCoV-19 (neuer Handelsname Vaxzevria) von AstraZeneca. Auch nach der Impfung mit Ad26.COV2.S werden zwar sehr selten, aber im Vergleich mit Plazebo vermehrt Thrombosen beobachtet. Wesentliche Unterschiede zu anderen bisher zugelassenen Impfstoffen gegen SARS-CoV-2 sind die einfachere Aufbewahrung und Verteilung des Impfstoffs und die nur einmalige Impfung – ein Vorteil besonders bei Massenimpfungen. Ob eine zweimalige Impfung nicht doch wirksamer ist, wird in einer noch laufenden Studie untersucht. Wichtige Fragen bleiben, wie bei anderen Impfstoffen gegen SARS-CoV-2, vorerst noch offen: Dauer der Immunität, Verhinderung der Virusübertragung ohne selbst zu erkranken, Wirksamkeit gegen neue Varianten von SARS-CoV-2 sowie Häufigkeit und Schweregrad seltener und/oder spät auftretender Nebenwirkungen.
Bei Ad26.COV2.S handelt es sich um einen genetischen Vektorimpfstoff, der auf einem nicht replizierenden, nicht pathogenen menschlichen Adenovirus (Typ 26) basiert. Der Vektor Ad26 wurde zuvor bereits von der Europäischen Arzneimittel-Agentur (EMA) für die Herstellung einer Vakzine gegen Ebola zugelassen und hatte sich in einer Phase-I/IIa-Studie hinsichtlich der Entwicklung einer humoralen und zellulären Immunität gegen SARS-CoV-2 als wirksam und sicher erwiesen (1). Das Prinzip der Vektorimpfstoffe haben wir im vergangenen November (2) und anlässlich der Zulassung von ChAdOx1 nCoV-19 (Vaxzevria) von AstraZeneca (Vektor = Schimpansen-Adenovirus) bereits ausführlich dargestellt (3). Der mittels rekombinanter DNA-Technologie hergestellte Virusvektor enthält ebenfalls einen für das SARS-CoV-2-Spike(S)-Glykoprotein kodierenden DNA-Abschnitt, der nach der Impfung vorübergehend durch körpereigene Zellen exprimiert und dem Immunsystem präsentiert wird. Die Immunantwort besteht dabei sowohl in der Bildung neutralisierender S-spezifischer Antikörper als auch in einer zellulären, gegen das S-Antigen gerichteten Immunreaktion. Zulassungsinhaber von Ad26.COV2.S ist Janssen Biotech Inc., eine Tochter des pharmazeutischen Unternehmers Johnson & Johnson.
Zulassung: Am 27.2.2021 wurde von der US-amerikanischen Food and Drug Administration (FDA) die Notfallzulassung (EUA = Emergency Use Authorization) und am 11.3.2021 von der EC die bedingte Zulassung für Ad26.COV2.S erteilt (4). Der Impfstoff ist für Menschen ab 18 Jahren zugelassen. Kontraindikationen sind Überempfindlichkeit gegen den Impfstoff oder gegen seine Hilfsstoffe. Ad26.COV2.S wird in den USA produziert; mit einer ersten Lieferung nach Europa ist frühestens Mitte April 2021 zu rechnen.
Beschreibung: Die rekombinanten Ad26.COV2.S-Vektorviren werden in PER.C6®-Zelllinien (transformierte fötale Retinoblastom-Zellen) vermehrt. Der fertige Impfstoff besteht aus einer Suspension von 5 x 1010 Vektorviren pro Impfdosis (0,5 ml), die i.m. verabreicht wird. Inaktive Hilfsstoffe sind unter anderen Zitronensäure-Monohydrat und Tri-Natriumcitrat-Dihydrat (als Puffer) sowie 2-Hydroxypropyl-β-Cyclodextrin (HPCD), Polysorbat 80 und eine geringe Menge Ethanol (als Stabilisatoren).
Ad26.COV2.S wird in Mehrfachdosen-Durchstechflaschen zu je fünf Dosen geliefert, muss lichtgeschützt bei 2°C bis 8°C – also bei Kühlschranktemperatur – gelagert werden und ist so 3 Monate haltbar. Nach Entnahme der ersten Dosis soll die Durchstechflasche maximal 6 Stunden im Kühlschrank oder maximal 2 Stunden bei Raumtemperatur (bis 25°C) gelagert werden. Ungeöffnete Durchstechflaschen können bis zu 12 Stunden bei Raumtemperatur gelagert werden. Ad26.COV2.S wird als Einmaldosis verimpft. Dieser Verabreichungsmodus ist logistisch deutlich einfacher als bei den anderen zugelassenen COVID-19-Impfstoffen – ein großer Vorteil bei Massenimpfungen.
Studienübersicht: Die Zulassungen durch die FDA und EMA stützen sich auf eine erste Zwischenanalyse (Stichtag 22. Januar 2021) von Daten einer großen, noch laufenden und leider noch nicht in einer Zeitschrift mit „peer review“ publizierten randomisierten plazebokontrollierten Phase-III-Studie („Study 3001“, ENSEMBLE; vgl. 6). Diese Studie wird in den USA, 6 Ländern Lateinamerikas (Mexiko, Peru, Kolumbien, Argentinien, Chile, Brasilien) und Südafrika durchgeführt (Rekrutierungsphase abgeschlossen). Insgesamt wurden 44.325 Probanden im Alter ≥ 18 Jahre eingeschlossen. Zu den Ausschlusskriterien zählten u.a. Erkrankungen und Situationen mit potenziellem Einfluss auf die erwünschte Immunantwort (z.B. Autoimmunerkrankungen, Hämodialyse, wiederholte systemische Glukokortikoid- oder antineoplastische Behandlungen in den vorausgehenden 6 Monaten; 8).
Die im Folgenden dargestellten Ergebnisse stammen, da sie noch nicht in einer Fachzeitschrift offiziell publiziert sind, aus dem European Public Assessment Report (EPAR; 4, 5).
Die „Per-Protocol“-Analyse zum Stichtag basierte auf den Daten von 39.321 Personen, die nach 1:1-Randomisierung entweder eine Dosis Ad26.COV2.S (n = 19.630) oder Plazebo (physiologische Kochsalzlösung; n = 19.691) i.m. erhalten hatten. Von den Probanden waren 34,6% im Alter ≥ 60 und etwa 20% im Alter 18 bis 40 Jahren. Primärer Endpunkt war ein ab 14 Tage (als „koprimärer“ Endpunkt später ergänzt: ab 28 Tage) nach der Impfung beginnender „mittel- bis schwergradiger“ Verlauf von COVID-19, bewertet durch ein sog. „Clinical Severity Adjudication Committee“. Definition der mittelschweren Erkrankung: Atemfrequenz ≥ 20/min oder Kurzatmigkeit oder verminderte Sauerstoffsättigung bei Raumluft oder klinische/radiologische Hinweise auf eine Pneumonie, tiefe Venenthrombose oder zwei der folgenden Kriterien: Fieber ≥ 38,0°C, Herzfrequenz ≥ 90/min, Schüttelfrost, Halsschmerzen, Husten, Krankheitsgefühl, Kopfschmerz, Myalgien, gastrointestinale Symptome, Geruchs- oder Geschmacksstörungen, Rötungen oder Hämatome an den Zehen. Definition der schweren Erkrankung: klinische Zeichen einer schweren Systemreaktion (Atemfrequenz ≥ 30/min, Herzfrequenz ≥ 125/min, Sauerstoffsättigung ≤ 93% bei Raumluft), signifikante renale, hepatische oder neurologische Funktionsstörung, Kreislaufschock, Notwendigkeit von Intensivbehandlung, nicht invasiver oder invasiver Beatmung, ECMO-Therapie (8). Die Infektion musste mittels PCR-Test aus einem Nasopharynx-Abstrich durch ein zentrales Studienlabor gesichert sein. Die mediane Nachbeobachtungszeit am Stichtag betrug 58 Tage.
Eine weitere laufende randomisierte, plazebokontrollierte Phase-III-Studie („Study 3009“; ENSEMBLE 2) untersucht an 30.000 Personen eine zweimalige Impfung mit Ad26.COV2.S (Impfabstand 57 Tage) gegenüber einer zweimaligen Impfung mit Plazebo. Diese Studie wird in den USA, Brasilien, Kolumbien, Südafrika, Philippinen sowie 5 europäischen Ländern (Belgien, Frankreich, Deutschland, Spanien und Großbritannien) durchgeführt und soll 2 Jahre laufen. Ob dieses Design durchgehalten werden kann, ist allerdings fraglich. Der pharmazeutische Unternehmer hat sich offenbar noch nicht endgültig auf das Konzept der Einmalimpfung festgelegt.
Die folgenden Ergebnisse stammen ausschließlich aus der „Study 3001“.
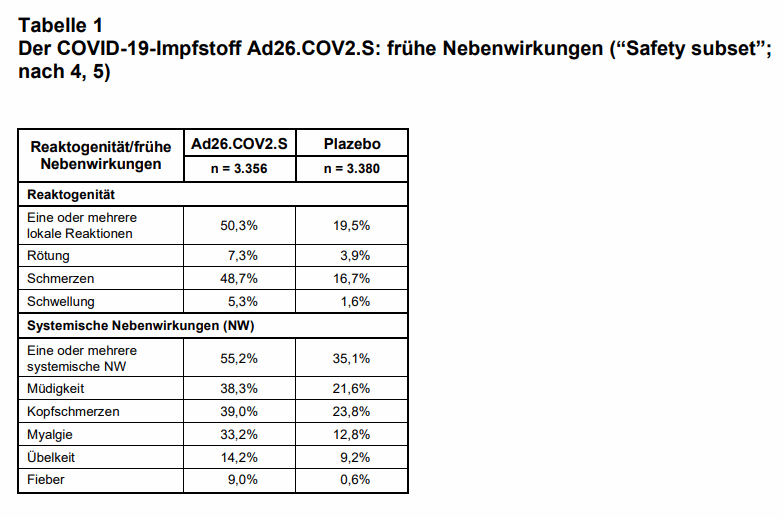
Sicherheit: Akute unerwünschte lokale oder systemische Wirkungen wurden in einem „Safety subset“ der Studienpopulation (n = 3.356 vs. n = 3.380) systematisch mit Hilfe eines elektronischen Tagebuchs erfasst und in den ersten 7 Tagen nach der Impfung bei 66% der Teilnehmer der Verumgruppe und bei 41,9% der Teilnehmer der Plazebogruppe registriert (vgl. Tab. 1). Die häufigsten Nebenwirkungen (NW) waren Schmerzen an der Injektionsstelle, Kopfschmerzen, Müdigkeit, Muskelschmerzen. Als „schwer“ („Grad 3“) wurden lokale NW bei 0,7% und systemische NW bei 1,8% der mit Verum Geimpften eingestuft. Damit löst Ad26.COV2.S eine etwa gleiche Reaktogenität aus wie ChAdOx1 nCoV-19 (Vaxzevria; zum Vergleich: nach Erstimpfung 71,3%, davon 8,1% mit Grad 3+4; vgl. 3), wobei sie generell milder und weniger häufig bei älteren Geimpften war.
Folgende Nebenwirkungen wurden in der gesamten Studienpopulation beobachtet:
Thromboembolische Ereignisse traten häufiger in der Verumgruppe auf (n = 11, davon 8 innerhalb von 28 Tagen nach Impfung) als in der Plazebogruppe (n = 4, davon alle innerhalb von 28 Tagen). Dies waren in der Verumgruppe 6 tiefe Beinvenenthrombosen (TVT), 4 Pulmonalembolien (PE) und eine Hirnvenenthrombose; in der Plazebogruppe gab es 2 TVT, eine PE und eine Hämorrhoidalthrombose (4, 5).
Der EPAR listet wenige Fälle von möglichen (aber teils fraglich Vakzin-induzierten) allergischen Reaktionen auf, die häufiger in der Verumgruppe als in der Plazebogruppe auftraten (Hautausschlag bei 24 vs. 16 Personen; Urtikaria 8 vs. 3). Es wird kein Fall von Anaphylaxie berichtet. In jeder Studiengruppe gab es einen Fall von Guillain-Barré-Syndrom sowie 3 vs. 2 „Fälle“ von Fazialisparese. Kausale Zusammenhänge mit der Impfung können aus diesen Beobachtungen aber nicht abgeleitet werden. Deshalb sind die im Rahmen der Pharmakovigilanz durchgeführten Routineaktivitäten für Impfstoffe gegen SARS-CoV-2 von größter Bedeutung (vgl. 7).
Wirksamkeit: Primärer Endpunkt: In der Verumgruppe erkrankten ab 14 Tage nach Impfung 116 (0,6%), in der Plazebogruppe 348 Probanden (1,8%) mittelschwer bis schwer an COVID-19. Hieraus errechnet sich eine Wirksamkeit von 66,9% (95%-Konfidenzintervall = CI: 59,03-73,40). Schwere Verläufe wurden ab 14 Tage nach Impfung bei 14 bzw. 60 Personen registriert (Wirksamkeit 76,7%) und ab 28 Tagen bei 5 bzw. 34 Personen (Wirksamkeit 85,4%). Die Ergebnisse in den meisten demographischen Subgruppen entsprachen weitgehend denen des primären Endpunkts. In der Altersgruppe ≥ 60 Jahre betrug die Wirksamkeit 76,3% (CI: 61,58-86,04), in der Altersgruppe 18 bis 59 Jahre 63,7% (CI: 53,87-71,58%).
Bis zum Stichtag wurden weniger Todesfälle in der Verumgruppe (n = 3; < 0,1%) registriert als in der Plazebogruppe (n = 16; 0,1%). Von den 16 Todesfällen in der Plazebogruppe wurden 6 als COVID-19-assoziiert eingestuft, dagegen keiner in der Verumgruppe. Um endgültige Aussagen zur Beeinflussung der COVID-19-Verläufe bzw. Gesamtmortalität durch Ad26.COV2.S machen zu können, sind jedoch mehr „Fälle“ bzw. größere Kohorten erforderlich.
Aus Subgruppenanalysen ergeben sich einige interessante Aspekte:
Virusvarianten: Zum Stichtag der Zwischenanalyse lagen erst für etwa zwei Drittel der COVID-19-Fälle S-Gen-Sequenzierungen für SARS-CoV-2 vor. In Südafrika (Gesamtpopulation: n = 4.969) dominierte mit 94,5% die Variante B.1.3.5 („südafrikanische Variante“). Die Wirksamkeit des Impfstoffs zur Verhinderung einer mäßig schweren oder schweren COVID-19-Erkrankung lag in Südafrika nur bei 39,6% (CI: 8,77-60,46) – ein möglicher Hinweis auf eine geringere Wirksamkeit des Impfstoffs bei der Variante B.1.3.5. In Brasilien (Gesamtpopulation: n = 6.725) betrug die Wirksamkeit von Ad26.COV2.S 66,6% (CI: 46,79-79,66) und war somit ähnlich wie in der gesamten Studienpopulation. Hier herrschte mit 69,4% die P.2-Variante vor („brasilianische Variante“). Es fand sich unter den beobachteten Infektionen bislang noch keine sequenzierte Variante B.1.1.7 („britische Variante“).
Im EPAR wird zu Recht betont, dass aufgrund der noch inkompletten und auf ausgewählte Patienten beschränkten Gen-Sequenzierungen (schwerere Verläufe, hohe Viruslast) verlässliche Aussagen zur Wirksamkeit von Ad26.COV2.S bei bestimmten Virusvarianten noch nicht möglich sind.
Wirksamkeit nach durchgemachter COVID-19-Erkrankung: 9,6% aller Probanden hatten zu Studienbeginn einen positiven SARS-CoV-2-Antikörperstatus. In dieser Subgruppe kam es zu sehr wenigen Fällen von COVID-19 (7 von 4.152 Probanden = 0,17%). Die „zusätzliche“ Wirksamkeit des Impfstoffs lag hier rechnerisch bei 28,5% (Verumgruppe: 3 von 2.122 = 0,14 %; Plazebogruppe: 4 von 2030 = 0,2%) – eine verlässliche Aussage ist aufgrund der wenigen Erkrankungen jedoch nicht möglich.
Ebenso wie bei allen anderen COVID-19-Impfstoffen sind auch zu folgenden weiteren Fragen derzeit keine Schlussfolgerungen möglich:
-
Dauer des Impfschutzes (hier über die Nachbeobachtungszeit von 2 Monaten hinaus),
-
Wirksamkeit gegen asymptomatische Infektionen und Virus-Übertragung (Transmission),
-
Einflüsse durch Virusmutationen, Änderungen im Pandemiegeschehen etc.,
-
Wirksamkeit und Sicherheit bei Kindern und Jugendlichen,
-
Wirksamkeit und Sicherheit bei Schwangeren,
-
Wirksamkeit gegen Langzeitfolgen nach COVID-19 („Late COVID“),
-
Wirksamkeit bei immunsupprimierten Personen,
-
Wirksamkeit bei spezifischen Komorbiditäten wie z.B. HIV/AIDS.
Immunogenität: Vorläufige Daten zur Immunogenität wurden aus 4 Studien erhoben („Study 3001“ sowie 3 Phase-I/II-Studien). In mehreren immunologischen Testverfahren zeigte sich dabei konsistent sowohl eine humorale als auch eine zelluläre, SARS-CoV-2-spezifische Immunantwort auf eine Einzeldosis mit 5 x 1010 Viruspartikeln. Diese Immunantwort war bei Älteren (≥ 60 Jahre) stärker ausgeprägt als bei Jüngeren (18 bis ≤ 55 Jahre). In letzterer Altersgruppe konnte eine über 3 Monate anhaltende Aktivität neutralisierender Antikörper dokumentiert werden. Aussagen über längere Zeiträume oder ältere Patientengruppen sind nicht möglich. Die Verabreichung einer 2. Impfdosis nach 8 Wochen führte zu einem 2,5- bis 3fachen Anstieg der Antikörper-Titer. Deshalb wird dieses Regime nun klinisch in einer weiteren Phase-III-Studie getestet („Study 3009“; s.o.).
Redaktionsschluss: 6.4.2021
Literatur
- Sadoff, J., et al.: N. Engl. J. Med. 2021, January 13. Link zur Quelle
- AMB 2021, 54, 85. Link zur Quelle
- AMB 2021, 55, 13. Link zur Quelle
- https://www.ema.europa.eu/en/medicines/human/ EPAR/covid-19-vaccine-janssen Link zur Quelle
- https://www.ema.europa.eu/en/ documents/product-information/ covid-19-vaccine-janssen-epar- product-information_en.pdf Link zur Quelle
- ENSEMBLE = A Study of Ad26.COV2.S for the Prevention of SARS-CoV-2-Mediated COVID-19 in Adult Participants: Link zur Quelle
- AMB 2021, 55, 01. Link zur Quelle
- ENSEMBLE Studienprotokoll: Link zur Quelle . Zugriff am 7.4.2021.