Zusammenfassung: Zwei aktuelle Studien legen dar, dass die Effektivität, insbesondere aber die klinische Sicherheit kardiovaskulärer Implantate vor ihrer Marktzulassung (noch) schlechter untersucht ist als die von Arzneimitteln. Die hohen Qualitätsanforderungen an Design und Durchführung von Arzneimittelstudien lassen sich nicht ohne weiteres auf Implantatstudien übertragen. In Anbetracht dieser Schwierigkeiten und des rasch wachsenden, sehr kostenintensiven und nicht selten auch ethisch sensiblen Bereichs ist es besonders wichtig, dass Studien zu Implantaten sorgfältiger prospektiv geplant werden und Qualitätskriterien erfüllen, die an die jeweilige Situation adaptiert sind. Nach der Marktzulassung könnten kontrollierte vollständige Register solcher Implantationen die vorhandenen Lücken im Qualitätsvergleich schließen im Sinne einer „Implantat-Vigilanz”. Die dazu notwendigen Informationen zur Häufigkeit von Reoperationen wegen Komplikationen oder technischer Defekte liegen (unveröffentlicht) bei den Kostenträgern.
Zwei aktuelle US-amerikanische Studien untersuchten die Qualität der Daten, die von den Herstellern kardiovaskulärer Implantate (Devices) in ihren Zulassungsanträgen an die FDA vorgelegt wurden, und kommen zu dem Schluss, dass die Marktzulassung oft trotz ungenügender Evidenz erfolgt. Die Untersuchungen umfassten unter anderen die Zulassungen folgender – heterogener – Gruppen von Implantaten: koronare und nicht-koronare Stents, endovaskuläre Grafts, intrakardiale Implantate (z.B. Okkludersysteme für Vorhofseptumdefekte), Herzschrittmacher und implantierbare Defibrillatoren, linksventrikuläre Unterstützungssysteme („Bridge-to-Transplant-Devices”).
Die erste Untersuchung (University of California, San Francisco) bewertete systematisch 123 öffentlich einsehbare ”Summaries of Safety and Effectiveness Data” (SSED) von 78 kardiovaskulären Implantaten, die von der FDA zwischen Januar 2000 und Dezember 2007 zugelassen wurden (1). Bereits aus diesen Zahlen geht hervor, dass jedem Zulassungsantrag im Mittel nur 1,6 Studien zugrunde liegen (bei 65% überhaupt nur eine, maximal fünf). Gerade mal 27% dieser Studien waren randomisiert, nur 14% verblindet. Von allen in diesen Studien untersuchten Endpunkten waren 88% Surrogatparameter, nur knapp mehr als die Hälfte hatten einen Vergleich mit einer Kontroll-Guppe, und 31% der Kontrollen waren retrospektiv. Die mittlere Patientenzahl lag bei 308 pro Studie, wobei 80% der SSED die Patientenzahl überhaupt nicht erwähnen. Die Autoren weisen auf den hohen Anteil von post-hoc-Analysen und die teilweise großen Diskrepanzen zwischen den Zahlen eingeschlossener und per-protocol analysierter Patienten hin.
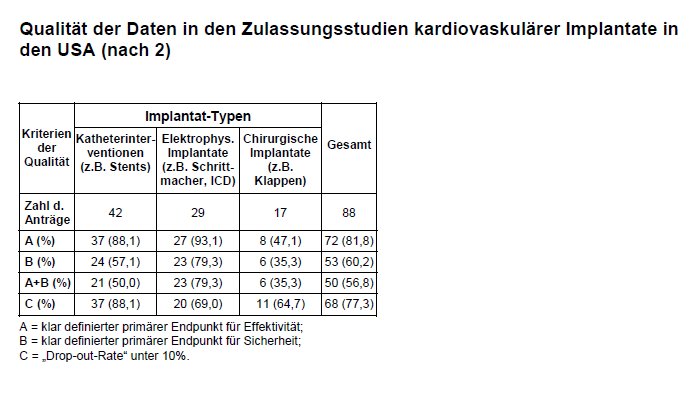
Die zweite – differenziertere – Untersuchung (2) stammt vom Medical Device Safety Institute des Beth Israel Deaconess Medical Center in Boston sowie Wissenschaftlern der Division of Cardiovascular Devices der FDA. Sie überprüfte alle Zulassungsanträge für kardiovaskuläre Implantate, über die die FDA von Januar 2000 bis Dezember 2007 zu entscheiden hatte. Den Autoren standen – im Unterschied zur erstgenannten Studie – nicht nur die öffentlich einsehbaren SSED, sondern die vollständigen Akten mit allen Studiendaten der bei der FDA eingereichten Zulassungsanträge zur Verfügung. Insgesamt wurden 88 Zulassungsanträge für kardiovaskuläre Implantate (davon 77,3% permanente Implantate) analysiert, denen 132 klinische Studien zugrunde lagen. Die mittlere Patientenzahl pro Studie lag bei 283, davon waren 232 Implantat-Empfänger. Angaben zu relevanten Komorbiditäten fanden sich nur in einem Teil der Studien (z.B. zu KHK nur in 51%, zu Diabetes nur in 37% und zu Nikotinabusus nur in 32% der Studien). Die Autoren überprüften die Studien auf ihre Datenqualität, wobei eine Studie als qualitativ hochwertig erachtet wurde, wenn folgende Kriterien erfüllt waren: A = klar definierter primärer Endpunkt für Effektivität; B = klar definierter primärer Endpunkt für Sicherheit mit jeweils spezifischen Analysezeitpunkten; C = „Drop-out-Rate” unter 10%. Diese Kriterien erfüllten nur A = 81,8%, B = 60,2% und C = 77,3% der Studien. Es wurden auch Implantate aus unterschiedlichen kardiovaskulären Anwendungsgebieten (s. Tab. 1) verglichen, wobei die Autoren in Anbetracht der sehr unterschiedlichen Implantate und Studiendesigns hier zur vorsichtigen Interpretation raten.
Aus den beiden Untersuchungen werden die großen Schwierigkeiten sichtbar, die mit der Evaluierung kardiovaskulärer Implantate verbunden sind:
· Bei den Implantationen handelt es sich in der Regel um permanente und irreversible Maßnahmen. Dagegen können Arzneimittel bei UAW abgesetzt werden.
· Bei manchen Implantaten handelt es sich um Therapieoptionen in Notfallsituationen als lebenserhaltende Maßnahme.
· Es ist schwierig und manchmal ethisch nicht vertretbar, Patienten für Implantatstudien zu randomisieren. Auch Verblindung ist in der Regel nicht möglich. Das Kriterium, ob eine Device-Studie randomisiert und verblindet ist oder nicht, ist daher nicht generell geeignet, die Qualität des Studiendesigns zu beurteilen. Dies trifft in besonderer Weise auf chirurgische Implantate zu.
· Die große Heterogenität der Implantate und ihrer Indikationen erfordert auch unterschiedliche Studiendesigns. Möglichst allgemeingültige Schemata – wie sie bei Arzneimittelstudien zur Qualitätssicherung gefordert werden – sind hier nicht sinnvoll.
· Studien mit sehr großen Patientenzahlen, wie sie bei kardiovaskulären Arzneimittelstudien üblich sind, sind bei Implantaten nicht praktikabel. Bei manchen Fragen ist man daher gezwungen, Surrogatparameter statt harter klinischer Endpunkte zu untersuchen, um überhaupt Daten zur Effektivität zu gewinnen.
· Lange Nachbeobachtungszeiten nach Implantationen werden häufig dadurch in Frage gestellt, dass von den Herstellern in immer kürzeren Zeitabständen Implantat-Innovationen angeboten werden und damit Vorgängermodelle obsolet werden.
Die FDA hat versprochen, Kriterien zu formulieren, die sich im Wesentlichen auf die Vorschläge von Kramer et al. (2) gründen. Künftig sollen Qualität und Zulässigkeit von Studien, auf die sich Implantat-Hersteller in ihren Zulassungsanträgen berufen, an diesen Kriterien gemessen werden (3):
· Klare Endpunkte für Sicherheit und Effektivität müssen vordefiniert sein.
· Der Zeitpunkt, zu dem ein Endpunkt evaluiert wird, muss vordefiniert sein.
· Bei kombinierten Endpunkten muss eine Gewichtung der einzelnen Komponenten vordefiniert sein.
· Patientenzahlen (insbesondere die Bilanz der Zahl eingeschlossener und analysierter Patienten) müssen dargelegt werden, um die Interpretierbarkeit der Ergebnisse zu gewährleisten.
Auf Kriterien, wie sie bei Arzneimittelstudien üblich sind (z.B. Randomisierung, Verblindung, möglichst lange Nachbeobachtung, kombinierte klinische Endpunkte inkl. Letalität) wurde aus oben genannten Gründen bewusst verzichtet.
Literatur
- Dhruva, S.S., et al.: JAMA 2009, 302, 2679. Link zur Quelle
- Kramer, D.B., et al.: Am. J. Ther. 2009, 17, 2. Link zur Quelle
- Miller. R.: theheart.org. 30. Dezember 2009. Link zur Quelle (Zugriff am 13.1.2010)