Zusammenfassung: Die klassischen Antiepileptika Valproinsäure, Carbamazepin, Phenobarbital/Primidon und Phenytoin können beim Menschen Entwicklungsauffälligkeiten verursachen. Hochgerechnet werden jedes Jahr in Deutschland mindestens 80-160 Kinder mit für Antiepileptika typischen Fehlbildungen geboren. Generell sind höhere Dosen bzw. eine Mehrfachtherapie riskanter für Störungen der Embryonalentwicklung als eine niedrig dosierte Monotherapie. Obwohl die klassischen Antiepileptika zu den am häufigsten verschriebenen, aber auch am besten untersuchten teratogenen Arzneimitteln in der Schwangerschaft gehören, ist die individuelle Abschätzung des Risikos immer noch schwierig. Betrachtet man die wichtigsten epidemiologischen Arbeiten der vergangenen Jahre (2, 4, 6, 11, 16, 17, 23, 25, 33), findet man Häufigkeitsangaben zwischen 3% und 11% für große Fehlbildungen unter antiepileptischer Monotherapie. Das entspricht ungefähr dem Zwei- bis über Vierfachen der jeweiligen Kontroll-Gruppen gesunder Schwangerer. Die unterschiedlichen Ergebnisse beruhen auf den jeweils schwerpunktmäßig erfassten Antiepileptika (Valproinsäure hat die höchste Teratogenität), auf der Definition „große Fehlbildungen”, dem Beobachtungszeitraum nach der Geburt sowie anderen Merkmalen des Studiendesigns und der Erfassungsqualität angeborener Anomalien. Noch verwirrender sind die Unterschiede bei den häufiger beobachteten kleinen Anomalien, Gesichts- und Fingerdysmorphien und den funktionellen ZNS-Störungen.
Jede 200. Schwangere leidet unter Epilepsie bzw. wird mit Antiepileptika behandelt. Verschiedene Aspekte sind in diesem Zusammenhang wichtig:
· Alle klassischen Antiepileptika haben ein embryotoxisches Potential.
· Ursprünglich wurde auch der Epilepsie selbst eine teratogene Wirkung zugeschrieben. Dies konnte jedoch weder für die Grunderkrankung noch für einzelne Krampfanfälle bestätigt werden.
· Sexualhormone können bei entsprechender Disposition krampffördernd (Östrogene) oder antikonvulsiv (Gestagene) wirken. Dies spielt z.B. bei zyklusabhängigen Krampfanfällen eine Rolle.
· Carbamazepin, Phenobarbital, Primidon, Phenytoin und Topiramat können über die Induktion des Zytochrom-P450-Enzymsystems zum Versagen oraler Kontrazeptiva (OK) und damit zu unerwünschten Schwangerschaften führen.
· Epilepsie und Antiepileptika können die Fertilität herabsetzen. Beispielsweise wird das mit anovulatorisch bedingter Fertilitätsminderung einhergehende polyzystische Ovar-Syndrom im Zusammenhang mit einer Temporallappen-Epilepsie und mit Valproat-Therapie diskutiert.
· Während der Schwangerschaft können häufiger Anfälle auftreten, weil der Wirkspiegel von Antiepileptika u.a. durch schlechtere Compliance und erhöhte Clearance sinkt.
Die wichtigsten Antiepileptika-assoziierten großen Fehlbildungen sind:
· Herzfehler (Häufigkeit 1,8%; 35),
· Lippen- und Gaumenspalten (Häufigkeit 1,7%; 35),
· Harnwegsanomalien, insbesondere Hypospadien,
· Skelettanomalien, z.B. Klumpfuß,
· Neuralrohrdefekte (Häufigkeit 1%-2% bei Carbamazepin und Valproat).
Zu den kleinen Anomalien bzw. Dysmorphien, dem sogenannten fetalen Antiepileptika-Syndrom, gehören:
· Mittelgesichtshypoplasie (kurze Nase, tiefliegender, breiter Nasenrücken bzw. Hypertelorismus, Epikanthus, lange Oberlippe),
· Auffälligkeiten der distalen Phalangen (kleine Nägel, kurze Finger-Endglieder, fingerähnlicher Daumen),
· Wachstumsrückstand,
· Mikrozephalie (insbesondere bei Phenytoin und Kombinationstherapie),
· Mentale Entwicklungsstörungen, Verhaltensauffälligkeiten sowie Hinweise auf Autismus-artige Symptome bei Valproinsäure.
Mentale Entwicklungsstörungen scheinen eher bei Kindern mit Mittelgesichtshypoplasie vorzukommen; die Angaben zur Häufigkeit bewegen sich im zweistelligen Prozentbereich (11, 24, 27, 28).
Als teratogene Mechanismen der klassischen Antiepileptika werden u.a. diskutiert:
· Folsäure-Antagonismus (z.B. 10),
· Mangel des für die Verstoffwechslung notwendigen Enzyms Epoxidhydrolase (29, 30),
· Hypoxie mit darauf folgender Reoxygenierung (3),
· Mangel an genetisch determinierter Expression von Retinsäure-Rezeptoren (8).
Zuerst wurden von Phenytoin Details zum Schädigungsmechanismus beschrieben: Zytochrom-P450-Enzyme metabolisieren Phenytoin zu reaktiven Arenoxiden, die sich im Embryo mit DNA oder Proteinen verknüpfen (39). Tierexperimentell konnte gezeigt werden, daß eine Phenytoin-Exposition die mRNA-Expression mehrerer Wachstumsfaktoren (z.B. TGFbeta, NT3 und WNT1) herabsetzt (26). Dass nur ein Teil der exponierten Kinder die typischen Anomalien zeigt, lässt sich wahrscheinlich mit dem disponierenden, genetisch determinierten Mangel an Epoxidhydrolase bei der Mutter und beim Embryo erklären, einem Enzym, das entwicklungstoxische Arenoxid-Metaboliten entgiftet. Dieses „eco-genetische” Zusammenspiel konnte schon vor über 20 Jahren am Beispiel eines dizygoten Zwillingpaares gezeigt werden: ein Zwilling war gesund, der andere zeigte die typischen Phenytoinauffälligkeiten – trotz gleichen intrauterinen Milieus.
Valproinsäure: Kinder mit dem sogenannten Valproinsäure-Syndrom unterscheiden sich vom einleitend beschriebenen Antiepileptika-Syndrom u.a. durch die Schädelform mit schmaler Stirn und durch die sich überkreuzenden schmalen Finger und Zehen und hyperkonvexen Nägel (19). An großen Fehlbildungen sind vor allem Neuralrohrdefekte typisch (20-40fach erhöhtes Risiko) sowie präaxiale Extremitätendefekte (31). Auch eine Kraniosynostose wurde im Zusammenhang mit Valproinsäure diskutiert (37). In neueren Studien werden mentale Entwicklungseinschränkungen durch Valproinsäure beschrieben und Häufigkeiten bis 20% angegeben. Verhaltensauffälligkeiten wie Aufmerksamkeitsdefizit, Hyperaktivität und autistische Symptome werden auch von anderen Autoren beschrieben (24, 40). Übererregbarkeit und andere neurologische Auffälligkeiten korrelieren laut einer weiteren Untersuchung mit der Valproinsäure-Konzentration bei der Geburt (18). Von einigen Autoren werden eine Schwellendosis von 1000 mg/d und eine Schwellenkonzentration im Serum von 70 µg/ml postuliert, unter der das teratogene Risiko erheblich geringer sei (17).
Der Verschluß des Neuralrohrs um Woche 6 herum ist ein komplexer Vorgang, zu dem u.a. die ungestörte Einwirkung verschiedener Wachstumsfaktoren Voraussetzung ist.
Tierexperimentelle Ergebnisse zeigten, dass teratogene Konzentrationen der Valproinsäure artspezifische Veränderungen der Genexpression bewirken, die wichtig für die normale Embryonalentwicklung sind. Dies betrifft u.a. Zellzyklus- und Apoptose-Gene. Außerdem wurden Veränderungen an Wachstumsfaktor-Genen wie z.B. BDGF (brain-derived growth factor), NGF (nerve growth factor) und den entsprechenden Rezeptoren beobachtet. Diese durch Valproinsäure induzierten Veränderungen der Genexpression werden möglicherweise durch relativen Folsäuremangel begünstigt bzw. durch entsprechende Zufuhr vermieden.
Der postulierte Zusammenhang von Valproinsäure und Autismus wird unterstützt durch Beobachtungen, daß Allelvarianten des HoxA1-Gens bei autistischen Personen gefunden wurden (30) und Valproinsäure in der Lage ist, die Expression von HoxA1 beim Embryo zu verändern. Auch unter Thalidomid waren autistische Symptome vermehrt gesehen worden und zwar dann, wenn der Embryo zum Zeitpunkt des Neuralrohr-Schlusses exponiert war.
Carbamazepin: Das ursprünglich als Carbamazepin-Syndrom beschriebene Bild (15) ist nicht eindeutig vom eingangs beschriebenen Antiepileptika-Syndrom zu unterscheiden. Jedoch muss bei Carbamazepin mit einem 10-20fach erhöhten Risiko für Neuralrohr-Defekte, insbesondere Spina bifida, gerechnet werden, d.h. ein bis zwei von 100 exponierten Kindern sind betroffen. Außerdem wurden gehäuft Hypospadien und Mikrozephalie beschrieben. Die teratogene Wirksamkeit von Carbamazepin scheint assoziiert zu sein mit einer deutlich erniedrigten Aktivität des Enzyms Epoxidhydrolase. Andererseits diskutieren einzelne Autoren eine protektive Wirksamkeit von Folsäure hinsichtlich des erhöhten Risikos für Neuralrohr-Defekte (9). Diese konnte jedoch weder bei Carbamazepin noch Valproinsäure eindeutig belegt werden.
Es gibt Hinweise darauf, dass Carbamazepin ähnlich wie Phenytoin einen Vitamin-K-Mangel mit resultierender Gerinnungsstörung beim Neugeborenen induzieren kann (12). Daher sollten Neugeborene zusätzlich zu den bei den Vorsorgeuntersuchungen üblichen Dosen in den ersten beiden Lebenswochen alle drei Tage 1 mg Vitamin K oral erhalten oder die erste Dosis direkt nach der Geburt i.m. zur verlässlicheren Resorption. Die Wirksamkeit einer Vitamin-K-Prophylaxe vor der Geburt bei der Mutter ist umstritten.
Phenobarbital und Primidon: In einer Gruppe von 46 Neugeborenen wurden bei 15% der Kinder auch für andere klassische Antiepileptika typische Gesichtsdysmorphien wie Epikanthus, Hypertelorismus, flache Nasenwurzel, aufwärtsgerichtete Nasenspitze etc. diagnostiziert (14). Außerdem hatten 24% der Kinder hypoplastische Fingernägel und drei von 16 nachuntersuchten Kindern Entwicklungsverzögerungen. Einige Untersucher betonen, daß Mischpräparate von Phenobarbital und Koffein das Fehlbildungsrisiko zusätzlich erhöhen (z.B. 33). Eine Störung des Steroid-, Vitamin-D- und Vitamin-K-Metabolismus wurde erörtert mit daraus resultierender Gerinnungsstörung und Blutungsneigung (12) sowie Hypokalziämie beim Neugeborenen. Andererseits wurde vermutet, daß eine unmittelbar präpartale Verabreichung, insbesondere bei Frühgeborenen, das Hirnblutungsrisiko senkt. Dies ließ sich aber nicht sicher belegen. Phenobarbital führt besonders in der Perinatalphase zu einer Aktivitätssteigerung der fetalen Leberenzyme. Das betrifft auch die glukuronidierenden Enzyme, die für Kopplung und Ausscheidung des Bilirubins verantwortlich sind. Wie bei Carbamazepin sollte das Neugeborene eine erweiterte Vitamin-K-Prophylaxe erhalten. Hyperirritabilität und Tremor können als Entzugssymptome in den Tagen nach der Geburt auftreten, wenn die Mütter in den letzten Monaten der Schwangerschaft täglich 60-300 mg Phenobarbital eingenommen haben. Zur Therapie mit Primidon in der Schwangerschaft liegen weit weniger dokumentierte Erfahrungen vor. Es sind jedoch ähnliche Effekte wie beim Phenobarbital zu erwarten.
Im Gegensatz zur langfristigen antikonvulsiven Anwendung sind Einzeldosen von Barbituraten (auch anderen als Phenobarbital) in (nicht zu empfehlenden) Schmerzmitteln oder im Rahmen einer Narkose wahrscheinlich nicht teratogen.
Phenytoin: Das teratogene Potential von Phenytoin ist seit 1964 bekannt (13). Ursprünglich wurden die im Zusammenhang mit Phenytoin beobachteten Anomalien als „fetales Hydantoin-Syndrom” bezeichnet (Übersicht bei 35). Nach heutiger Ansicht unterscheidet es sich jedoch nicht signifikant vom eingangs beschriebenen Antiepileptika-Syndrom. Einschränkungen der kognitiven Entwicklung wurden auch nach intrauteriner Phenytoin-Exposition gehäuft beobachtet (36, 38). Konflikte bei der späteren geschlechtsspezifischen Identitätsentwicklung werden an anderer Stelle diskutiert (5); sie sind jedoch bisher nicht von anderen Untersuchern bestätigt.
Einige Veröffentlichungen beschäftigten sich mit dem möglichen Risiko einer transplazentaren Karzinogenese durch Phenytoin. Zwölf pränatal exponierte Kinder mit neuroektodermalen Tumoren wurden beschrieben, sechs davon mit Neuroblastomen (Übersicht in 1). Die Fallzahlen sind jedoch zu klein, um einen kausalen Zusammenhang als wahrscheinlich anzunehmen.
Bei Neugeborenen kann es nach Phenytoin-Exposition ähnlich wie nach Barbituraten zu Gerinnungsstörungen durch Vitamin-K-Mangel kommen (Übersicht bei 1). Wie bei Carbamazepin beschrieben, sollte das Neugeborene eine erweiterte Vitamin-K-Prophylaxe erhalten.
Diazepam und Clonazepam: Frühere Veröffentlichungen haben ein erhöhtes Risiko für Lippen-Kiefer-Gaumen-Spalten unter Diazepam beschrieben. Dies wurde von anderen Autoren nicht bestätigt (32). Gleiches gilt für andere Fehlbildungen, wie z.B. Inguinalhernien. In einer Fallserie mit acht Kindern, deren Mütter während der gesamten Schwangerschaft Arzneimittelabusus mit täglich mindestens 30 mg Diazepam und mindestens 75 mg Oxazepam betrieben hatten, hatten alle Kinder Gesichtsdysmorphien, einige außerdem eine Mikrozephalie sowie postpartal toxische Symptome (Apnoe) und Entzugserscheinungen (21). Später wurden unterschiedlich ausgeprägte mentale Retardierungen, Konzentrationsstörungen und Hyperkinesien beobachtet. Den Autoren dieser Falldarstellungen wurde jedoch vorgehalten, Art und Umfang der Exposition nicht ausreichend recherchiert und Differenzialdiagnosen, wie z.B. ein Zellweger-Syndrom, nicht ausgeschlossen zu haben. In Nachfolgeuntersuchungen wurde bei den dann etwa 18 Monate alten Kindern eine Besserung der Symptome festgestellt (20). Eine Metaanalyse aller verwertbaren Kohortenstudien zur Benzodiazepin-Exposition erbrachte keine Auffälligkeiten. Die in derselben Publikation analysierten Fall-Kontroll-Studien fehlgebildeter Kinder ergab hingegen ein erhöhtes Fehlbildungsrisiko, z.B. für isolierte Mundspalten (7).
Alle bis heute vorliegenden Daten zusammengefasst scheint kein den klassischen Antiepileptika vergleichbares teratogenes Risiko vorzuliegen. Gesichert sind hingegen die funktionellen Störungen beim Neugeborenen, wenn unter der Geburt Benzodiazepine hochdosiert verabreicht wurden oder wenn über längere Zeiträume, das letzte Schwangerschaftsdrittel inbegriffen, regelmäßig 15-20 mg und mehr Diazepam oder analoge Mengen anderer Benzodiazepine eingenommen wurden. Einerseits muss nach hohen Dosen sub partu mit Atemdepression gerechnet werden, andererseits kann nach andauernder Exposition eine Entzugssymptomatik mit Unruhe, Tremor, Muskelhypertonus, Erbrechen und Durchfall auftreten wie nach Opiaten. Auch zerebrale Krampfanfälle in der Neonatalphase sind möglich und ein Wochen bis Monate anhaltendes „Floppy-infant-Syndrom” mit Muskelschlaffheit, Lethargie, Störungen der Temperaturregulation und Trinkschwäche (Übersicht bei 1). Durch Akkumulation im Feten können im Einzelfall bereits tägliche Dosen von 6 mg Diazepam beim Neugeborenen zu klinischen Symptomen führen. Benzodiazepine können – unter der Geburt verabreicht – Bilirubin aus der Albuminbindung verdrängen und zumindest theoretisch einen Icterus neonatorum verstärken.
Clonazepam ähnelt chemisch-strukturell dem Diazepam. Zur intrauterinen Exposition liegen kleinere Fallzahlen vor, u.a. eine Kohortenstudie mit etwa 50 Schwangeren (22). Ein erhebliches teratogenes Risiko ließ sich ebenso wenig nachweisen wie bei anderen Benzodiazepinen. Neonatal ist mit den gleichen Symptomen wie bei Diazepam zu rechnen.
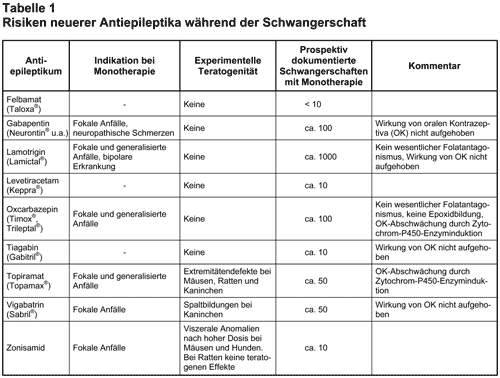
Neuere Antiepileptika: Zu den neueren Antiepileptika zählen Felbamat, Gabapentin, Lamotrigin, Levetiracetam, Oxcarbazepin, Tiagabin, Topiramat, Vigabatrin, Zonisamid. Zu Lamotrigin liegen mit etwa 1000 die meisten prospektiven Verlaufsbeobachtungen zu einer Monotherapie vor (Lamotrigine Pregnancy Registry – GlaxoSmithKline, 2004; New AEDs in Pregnancy-UK Register Belfast; Pharmakovigilanzzentrum Embryonaltoxikologie Berlin). Diese deuten bislang nicht auf spezifische teratogene Effekte hin, so dass – vorsichtig ausgedrückt – möglicherweise ein geringeres Risiko als bei den klassischen Antiepileptika besteht. Tab. 1 gibt einen Überblick zu den Größenordnungen an prospektiv nachverfolgten Schwangerschaften unter Monotherapie sowie den Erkenntnissen zur experimentellen Teratogenität der neueren Antiepileptika.
Empfehlungen zur antiepileptischen Therapie in der Schwangerschaft:
· Besteht jahrelang Anfallsfreiheit, sollte vor Planung einer Schwangerschaft die Beendigung der antiepileptischen Behandlung erörtert werden.
· Aufgrund des im Vergleich zu den anderen Antiepileptika hohen teratogenen Risikos von Valproinsäure sollte diese bei Planung einer Schwangerschaft oder besser im gesamten reproduktionsfähigen Alter bei Frauen nur noch dann verordnet werden, wenn andere Antiepileptika unzureichend wirken. Valproinsäure sollte nicht für andere Indikationen, z.B. als Phasenprophylaktikum bei bipolaren Erkrankungen, verwendet werden, weil hierfür besser verträgliche Psychopharmaka zur Verfügung stehen (34).
· Eine Monotherapie ist anzustreben. Die Tagesdosis des Antiepileptikums sollte auf 3-4 Einzeldosen verteilt und Retardpräparate bevorzugt werden.
· Eine Folsäure-Prophylaxe mit 5 mg/d sollte ab Planung der Schwangerschaft bis Woche 10 erfolgen.
· Eine Untersuchung mit hochauflösendem Ultraschall sollte zur Bestätigung der normalen fetalen Entwicklung angeboten werden.
· Bei Antiepileptika mit Vitamin-K-antagonistischer Wirkung sollte das Neugeborene in den ersten beiden Lebenswochen oral alle 3 Tage 1 mg Vitamin K erhalten oder die erste Dosis direkt nach der Geburt i.m. zur verlässlicheren Resorption.
· Monatliche Konzentrationsbestimmungen im mütterlichen Serum, insbesondere bei Antiepileptika mit hoher Eiweißbindung, ermöglichen eine adäquate Dosisanpassung in der Schwangerschaft.
Literatur
- Briggs, G.G., et al.: Drugs in Pregnancy and Lactation. 6th edition. Williams and Wilkins, Baltimore 2002.
- Canger, R., et al.: Epilepsia 1999, 40, 1231.
- Danielsson, B.R., et al.: Teratology 1997, 56, 271.
- Dean, J.C., et al.: J. Med. Genet. 2002, 39, 251.
- Dessens, A.B., et al.: Arch. Sex. Behav. 1999, 28, 31.
- Diav-Citrin, O., et al.: Neurology 2001, 57, 321.
- Dolovich, L.R., et al.: Brit. Med. J. 1998, 317, 839.
- Gelineau-van Waes, J., et al.: Teratology 1999, 59, 23.
- Hernandez-Diaz, S., et al.: Am. J. Epidemiol. 2001, 153, 961.
- Hernandez-Diaz, S., et al.: N. Engl. J. Med. 2000, 343, 1608.
- Holmes, L.B., et al.: N. Engl. J. Med. 2001, 344, 1132.
- Howe, A.M., et al.: Epilepsia 1999, 40, 980.
- Janz, D., et al.: German Med. Monogr. 1964, 9, 201964, 9, 20.
- Hanson, J.W., und Smith, D.W.: J. Pediatr. 1975, 87, 285.
- Jones, K.L., et al.: N. Engl. J. Med. 1989, 320, 1661.
- Kaaja, E., et al.: Neurology 2003, 60, 575.
- Kaneko, S., et al.: Epilepsy Res. 1999, 33,145.
- Koch, S., et al.: Acta Paediatr. 1996, 85, 739.
- Kozma, C.: Am. J. Med. Genet. 2001, 98, 168.
- Laegreid, L., et al.: Neuropediatrics 1992, 23, 60.
- Laegreid, L., et al.: J. Pediatr. 1989, 114, 126.
- Lin, A.E., et al.: Birth Defects Research, Part A Clin. Mol. Teratol. 2004, 70, 534.
- Matalon, S., et al.: Reprod. Toxicol. 2002, 16, 9.
- Moore, S.J., et al.: J. Med. Genet. 2000, 37, 489.
- Morrow, J.I., und Craig, J.J.: Expert Opin. Pharmacother. 2003, 4, 445.
- Musselman, A.C., et al.: Reprod. Toxicol. 1994, 8, 383.
- Ornoy, A., und Cohen, E.: Arch. Dis. Child. 1996, 75, 517.
- Orup, H.I. Jr., et al.: Orthod. Craniofac. Res. 2003, 6, 2.
- Raymond, G.V., et al.: Teratology 1995, 51, 55.
- Rodier, P.M.: Neuroteratology of autism. In: Slikker, W., und Chang, L. (Hrsg.): Handbook of developmental neurotoxicology. Academic Press, New York 1998. S. 661.
- Rodriguez-Pinilla, E., et al.: Am. J. Med. Genet. 2000, 90, 376.
- Rosenberg, L., et al.: N. Engl. J. Med. 1983, 309, 1282.
- Samren, E.B., et al.: Ann. Neurol. 1999, 46, 739.
- Schaefer, C., et al.: Arzneiverordnung in Schwangerschaft und Stillzeit. 6. Aufl., Urban & Fischer, München 2001.
- Schardein, J.L.: Chemically Induced Birth Defects. 3. Aufl., Marcel Dekker, New York 2000.
- Scolnik, D., et al.: JAMA 1994, 271, 767.
- Shepard, T.H., et al.: Teratology 2002, 65, 153.
- Vanoverloop, D., et al.: Neurotoxicol. Teratol. 1992, 14, 329.
- Wells, P.G., et al.: Reactive intermediates. In: Kavlock, R.J., und Daston, G.P. (Hrsg.): Drug toxicity in embryonic development. Vol. 1, Handbook of experimental pharmacology. Vol. 124/I. Springer, Heidelberg 1997. S. 451.
- Williams, G., et al.: Dev. Med. Child. Neurol. 2001, 43, 202.