Zusammenfassung: Die chronisch obstruktive Lungenerkrankung (COPD) wird als eine Krankheit definiert, bei der eine abnorme Entzündungsreaktion auf inhalative Noxen zu bronchialer Hypersekretion und obstruktiver Bronchiolitis führt. Die Folgen sind erhebliche strukturelle Veränderungen der Lunge mit Abnahme ihrer Funktion sowie allgemeine körperliche Krankheitssymptome. Proinflammatorische Zytokine unterhalten die Entzündung und bieten möglicherweise Ansätze für eine spezifische und effektivere Therapie. Die medikamentöse Therapie der COPD ist in erster Linie an den Symptomen orientiert; kein Arzneimittel kann bisher die kontinuierliche Verschlechterung der Lungenfunktion aufhalten oder die Letalität senken. Die Tabakentwöhnung ist die wirksamste Maßnahme zur langfristigen Stabilisierung der Lungenfunktion. Seit unserer Übersicht im Jahre 2003 haben sich durch neue Befunde zusätzliche Perspektiven ergeben. Im Februar 2006 ist eine nationale Versorgungsleitlinie und 2007 eine aktualisierte Leitlinie der Deutschen Gesellschaft für Pneumologie erschienen.
Die COPD ist hinsichtlich Morbidität und Letalität eine sehr bedeutende Erkrankung. Sie wird bei steigender Inzidenz bis zum Jahr 2020 auf dem dritten Platz der Todesursachenstatistik in Europa prognostiziert (1). Der Verlauf ist chronisch und irreversibel mit progredienter Atemwegsobstruktion und Dyspnoe auf dem Boden einer chronischen Bronchitis mit oder ohne Lungenemphysem. Im Gegensatz zum Asthma bronchiale ist die Atemwegsobstruktion nach Gabe von Bronchodilatatoren und/oder Kortikosteroiden nicht vollständig reversibel (2).
Die wichtigste Ursache der COPD ist eine chronische, abnorme Entzündungsreaktion der Lunge auf inhalative Noxen. Die Schleimhautirritation führt zur Hyperplasie der bronchialen Becherzellen mit vermehrter Sekretproduktion und zur Metaplasie des Epithels in den zentralen Atemwegen. Durch aktivierte Alveolarmakrophagen wird eine lokale Inflammationskaskade in Gang gesetzt. Dabei werden Proteasen, Fibroblasten, neutrophile Granulozyten und CD8-positive T-Lymphozyten stimuliert und vermehrt Tumor-Nekrose-Faktor alpha (TNF alpha), Interleukin 8 (IL-8), Leukotrien-B4 (LTB-4) und andere Chemokine sezerniert (s. Abb. 1). Bereits geschädigte Bronchial- und Alveolarepithelzellen unterhalten die Entzündung (3). Hierdurch und als Folge von peribronchialer Fibrose, Hypertrophie der bronchialen Muskelzellen und Hypersekretion von Schleim entsteht eine chronische obstruktive Bronchiolitis (small airway disease) mit erhöhtem Atemwegswiderstand. Ein Ungleichgewicht von Proteasen und Anti-Proteasen führt zum irreversiblen Untergang von Alveolarsepten und zum Emphysem. Die genetischen und/oder zellulären Mechanismen der unterschiedlichen COPD-Phänotypen (Pink puffer, Blue bloater) sind Gegenstand der Forschung. Es ist auch nicht geklärt, warum nur eine Minderheit der Raucher (25%) im Verlauf von 25 Jahren eine COPD entwickelt (4, 5).
Tabakrauch ist der wichtigste Risikofaktor für die COPD. Wegen dieser Assoziation wird die COPD in den Leitlinien 2007 erstmals als eine vermeidbare Krankheit charakterisiert (6). In der aktualisierten Definition wird sie jetzt als multifaktoriell verstanden, bei der die Atemwege der Ausgangspunkt für eine systemische Erkrankung sind, die mit Dyspnoe, Gewichtsverlust, Muskelschwäche, Osteoporose, Depression und endokriner Dysfunktion verläuft.
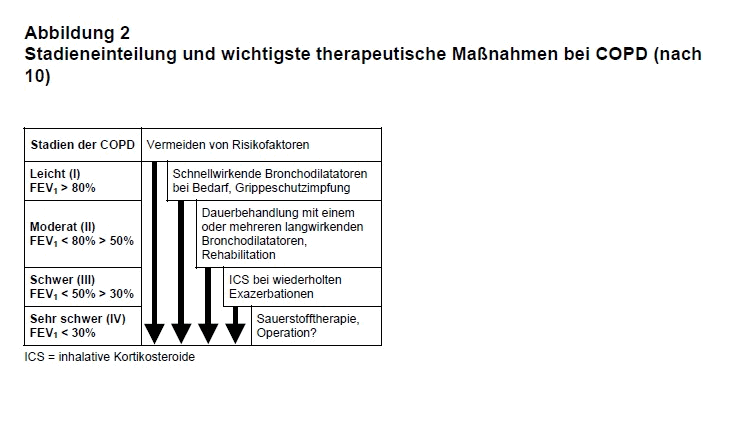
Bei der Einteilung der COPD nach GOLD (Global initiative for chronic Obstructive Lung Disease) werden vier Schweregrade unterschieden (s. Abb. 2; 7). Das frühere Stadium 0 (Patienten mit chronischem Husten und Auswurf bei normaler Spirometrie) gilt nicht mehr als risikobehaftete Vorstufe der COPD. Entgegen der bisherigen Annahme entwickeln Betroffene nämlich nicht häufiger eine klinisch manifeste COPD als der Durchschnitt der Bevölkerung. Ferner wurde die Unterteilung des Stadiums II in IIA und IIB aufgegeben. Hinsichtlich der Prognose ist die 50%-Schwelle des forcierten exspiratorischen Ein-Sekunden-Volumens (FEV1) besonders relevant, denn unterhalb dieses Werts kommt es deutlich häufiger zu Exazerbationen mit wesentlich höherem Morbiditäts- und Letalitätsrisiko.
Zur Prognoseeinschätzung wurde der BODE-Index in die Leitlinien aufgenommen. Er erfasst Body-Mass-Index, FEV1 (Airflow Obstruction), Dyspnoeeinstufung und körperliche Belastbarkeit (Exercise capacity), beurteilt im Sechs-Minuten-Gehtest (s. Tab. 1). Der BODE-Score sagt das Letalitätsrisiko bei COPD wesentlich zuverlässiger voraus als das FEV1 allein. Nach einer Studie von Celli, B.R., et al. ist die Zunahme des BODE-Scores um einen Punkt mit einer Hazard ratio (HR) für Tod aus allen Ursachen von 1,34 (95%-Konfidenzintervall = CI: 1,26-1,42; p < 0,001) assoziiert. Für Tod aus respiratorischen Ursachen beträgt die HR 1,62 (CI = 1,48-1,77; p < 0,001) für jeden zusätzlichen Punkt. Die höchste BODE-Kategorie (7-10 Punkte) ist mit einer Letalität von 80% nach 52 Monaten assoziiert (8).
Medikamentöse Therapie: Die medikamentöse Therapie der COPD soll die Symptome lindern, die Belastbarkeit fördern und Exazerbationen reduzieren. Sie kann aber bisher nicht die kontinuierliche Abnahme der Lungenfunktion aufhalten oder die Letalität senken. Bei Rauchern ist im Vergleich zu Gesunden mit einer schnelleren Abnahme des FEV1 (um > 50 ml/Jahr) zu rechnen. Durch Aufgabe des Rauchens kann die Progression zwar gebremst, aber nicht vollständig aufgehalten werden. Dennoch ist dies die einzig wirksame Maßnahme, um den Verlauf der Krankheit positiv zu beeinflussen (9).
Am Stufenschema der medikamentösen Therapie bei stabiler COPD hat sich in den letzten Jahren und in den Leitlinien 2007 nichts grundlegend geändert (s. Abb. 2; 10). Wir haben früher bereits ausführlich darüber berichtet (11). In allen Stadien ist das Vermeiden von Risikofaktoren und Noxen essenziell. Die Grippe- und die Pneumokokken-Schutzimpfung wird angeraten. Eine pulmonale Rehabilitation sollte fester Bestandteil jeder Begleitung eines COPD-Kranken sein (vgl. 12).
Im Stadium I sind weiterhin kurz wirksame (< 6 h) Bronchodilatatoren wie Fenoterol oder Salbutamol indiziert. Ab GOLD-Stadium II haben sich lang wirkende Beta2-Sympathikomimetika wie Formoterol oder Salmeterol (> 12 h) als effektiver erwiesen als Substanzen mit kurzer Wirkdauer. Zudem sind sie für den Patienten bequemer anzuwenden. Darüber hinaus ist belegt, dass die kombinierte Gabe eines lang wirkenden Beta2-Sympathikomimetikums mit dem ebenfalls lang wirkenden Anticholinergikum Tiotropium die Lungenfunktion deutlicher verbessert als eine Monotherapie mit den Einzelsubstanzen (13). Die Wirkung von Tiotropium hält länger an (bis zu 36 h) als die von Ipratropium (< 8 h). Eine randomisierte, doppeltblinde, multizentrische Untersuchung über vier Jahre an fast 6 000 Patienten hat gezeigt, dass Tiotropium - zusätzlich zur Standardtherapie - subjektiv zwar zu einer deutlichen Besserung führt („Lebensqualität”, Zahl der Exazerbationen), den langsamen Rückgang des FEV1 aber nicht aufhalten kann (14). In dieser Studie nahm die Zahl kardiovaskulärer Komplikationen unter der Behandlung mit Tiotropium nicht zu. Jedoch gibt es Hinweise in diese Richtung (15), die weitere Untersuchungen dringend erforderlich machen.
Inhalierbare Kortikosteroide (ICS) sind erst indiziert, wenn das FEV1 auf < 50% sinkt und der Patient pro Jahr mindestens eine Exazerbation erleidet, die Kortikosteroide und/oder Antibiotika erfordert (GOLD-Stadium III). Die inhalative Therapie - kombiniert Salmeterol plus Fluticason - bei schwerer COPD kann zwar die langsame Progression auch nicht aufhalten und die Gesamtletalität nicht mindern, aber das allgemeine Wohlbefinden (die „Lebensqualität”) bessern und die Zahl der Exazerbationen reduzieren. Das zeigt eine viel zitierte, u.a von der Herstellerfirma GlaxoSmithKline unterstützte, kontrollierte, doppeltblinde Untersuchung an mehr als 6 000 Patienten (16). Aus einer aktuellen Metaanalyse zu ICS ergaben sich allerdings Hinweise für ein erhöhtes Pneumonierisiko ohne negativen Einfluss auf die Ein-Jahres-Letalität (17). Diese Beobachtung wurde bisher noch nicht durch kontrollierte Studien belegt, sollte aber motivieren, die niedrigste noch effektive ICS-Dosis zu ermitteln, um unerwünschte Arzneimittelwirkungen (UAW) zu minimieren.
Im Stadium IV kann eine Langzeit-Sauerstofftherapie bei geeigneten Patienten die Symptome lindern (18). Eine OP zur Reduktion des Lungenvolumens oder eine Lungentransplantation kann bei geeigneten Patienten als ultima ratio erwogen werden. Bei Patienten mit schwerer Exazerbation und respiratorischem Versagen bei Hyperkapnie ist heute die nicht-invasive Maskenbeatmung (NIV) Standard. Dadurch kann eine Intubation häufig vermieden werden. In einer 2008 zur NIV herausgegebenen Leitlinie wird hierzu präzise Stellung genommen (19).
Sekretolytika sind als supportive Therapie seit jeher umstritten. Acetylcystein (ACC) liefert die Aminosäure Zystein für das körpereigene Antioxidans Glutathion, das dem „oxidativen Stress” bei COPD entgegenwirken soll. Anders als in früheren Metaanalysen konnten Exazerbationen durch ACC (600 mg/d) nicht reduziert bzw. Verschlechterungen der Lungenfunktion nicht aufgehalten werden, wenn die Patienten auch gleichzeitig ein ICS erhielten, so wie es heute bei fortgeschrittener COPD Standard ist (20-22). Ein positiver Einfluss von ACC auf die Verschlechterung der Lungenfunktion konnte bisher nur bei Lungenfibrose in einer Dosis von 1800 mg/d gezeigt werden. Nur unter dieser hohen Dosierung werden in der Lunge Glutathionkonzentrationen wie bei Lungengesunden erreicht. Studien mit dieser Dosis bei COPD liegen bisher nicht vor.
Kortikosteroide (z.B. Prednisolon 20-60 mg/d i.v. oder oral) sind den akuten Exazerbationen vorbehalten und sollten nicht länger als zwei Wochen gegeben werden. Bei schweren Exazerbationen können auch höhere Dosen erforderlich sein. Sie verkürzen den stationären Aufenthalt und vermindern Rezidive, verbessern aber nicht anhaltend die Lungenfunktion. Sie können nach bis zu zweiwöchiger Gabe ohne Ausschleichen abgesetzt werden. Eine Dauertherapie in niedriger Dosis ist generell nicht zu empfehlen, denn sie birgt das Risiko häufigerer Infektexazerbationen und senkt nicht die Letalität. Ist bei einzelnen Patienten durch keine andere Maßnahme eine Besserung des FEV1 – vor allem des Allgemeinbefindens – zu erreichen, kann sie unter kritischem Abwägen jedoch gerechtfertigt sein.
Theophyllin ist ein unspezifischer Phosphodiesterase-Inhibitor mit relativ schwacher bronchodilatatorischer Wirkung und geringer therapeutischer Breite. Es ist aber additiv wirksam zusammen mit Beta2-Sympathikomimetika und ICS. Typische UAW sind Übelkeit, Herzrhythmusstörungen und Senkung der Krampfschwelle. Eine Dauermedikation sollte deshalb nur solchen Patienten verordnet werden, die von der Therapie tatsächlich profitieren. Möglicherweise wirkt Theophyllin in subtherapeutischen Dosen (Serumkonzentration < 10 mg/l) auch antiinflammatorisch, denn es sinken die Zahl der neutrophilen Granulozyten sowie die Konzentrationen von IL-8, neutrophiler Elastase und Myeloperoxidase (23).
Antibiotische Therapie: Sie ist nicht indiziert bei einfacher, nicht obstruktiver Bronchitis. Etwa ein Drittel der Infektexazerbationen sind viraler Genese. Bei klinischen Hinweisen auf eine bakterielle Infektion (eitriges Sputum) und progredienter Dyspnoe sowie bei bereits schwerer Einschränkung der Lungenfunktion und anamnestisch häufigen Exazerbationen sollte die Indikation großzügiger gestellt werden. Die Erreger sind überwiegend Pneumokokken, Haemophilus influenzae und Moraxella catarrhalis, die in der Regel gegen Betalaktam-Antibiotika, Cefalosporine, neuere Makrolide und Fluorchinolone sensibel sind. Ciprofloxacin wird als Monotherapie wegen zunehmender Resistenzen gram-positiver Erreger nicht mehr empfohlen. Bei einem FEV1 < 35% muss häufiger mit gram-negativen Keimen und Pseudomonas spp. gerechnet werden, die einer speziellen Antibiotikatherapie bedürfen (24).
Neue Therapieansätze: Immunmodulatorische Substanzen können in die inflammatorischen Prozesse eingreifen, die sich im fortgeschrittenen Stadium der COPD eigendynamisch, d.h. unabhängig von der ursprünglichen Noxe, unterhalten (s. Abb. 2). Verschiedene Chemokine aktivieren die Granulozytenmigration in die Alveolen, u.a. IL 8 und LTB4. Elastin-abbauende Proteasen führen zu strukturellen Veränderungen der Lunge mit Zerstörung der Alveolarwand und Emphysembildung. Die Antagonisierung dieser Chemokine durch monoklonale Antikörper könnte möglicherweise die Kaskade der Umbauvorgänge im Lungenparenchym reduzieren. Antikörper und Rezeptorblocker gegen IL-8 sind in Entwicklung (25). Sie sind möglicherweise eine zukünftige Therapieoption für die Patienten, die sehr hohe Entzündungsparameter und IL-8-Konzentrationen im Serum oder Sputum haben und die unter häufigen Exazerbationen leiden. Inhibitoren der p38 mitogen-aktivierten Proteinkinase könnten die Bildung inflammatorischer Zytokine modulieren. In vitro hemmen sie die Freisetzung von TNF alpha aus Lungenmakrophagen. Hohe Konzentrationen von TNF alpha sind bei COPD-Patienten mit verstärktem Muskelabbau und protrahiertem Gewichtsverlust korreliert. Ein monoklonaler Antikörper gegen TNF alpha ist z.B. als Infliximab (Remicade®) im Handel. Die Wirksamkeit dieser teuren Substanz ist aber bei COPD nicht belegt. Auch für Leukotrien-Antagonisten ist die Datenlage bisher negativ, anders als in der Therapie des Asthma bronchiale (20). Aufgrund der unspezifischen Phosphodiesterase(PDE)-Hemmung von Theophyllin wurden spezifischere PDE-4-Inhibitoren entwickelt, die in den Stoffwechsel von c-AMP in glatten Muskel- und in Entzündungszellen eingreifen und bronchodilatatorische und antiinflammatorische Effekte haben. Sie haben aber erhebliche UAW (26). So wurde Cilomilast von der amerikanischen Zulassungsbehörde wegen Sicherheitsbedenken bei unzureichender Wirksamkeit abgelehnt. Der Zulassungsantrag für Roflumilast wurde 2005 zurückgezogen wegen schwerer gastrointestinaler UAW. Wir haben seinerzeit darüber berichtet (27). Makrolide haben nicht nur antibiotische, sondern in vitro auch antiinflammatorische Effekte (28). In antibiotisch subtherapeutischen Dosen senken sie die Konzentrationen von IL-8 und IL-6 und hemmen MMP-9, eine elastolytische Matrixmetalloprotease der Alveolarmakrophagen, die besonders für das „Capillary leakage” beim Lungenversagen verantwortlich ist. In einer kürzlich erschienenen Studie reduzierte eine niedrig dosierte (250 mg/d), perorale Langzeittherapie mit Erythromycin (12 Monate) die Zahl der Klinikaufenthalte und verlängerte die Exazerbationsintervalle (29). Die Ergebnisse einer weiteren, großen Studie zu dieser Therapie werden noch in diesem Jahr erwartet. Die Vorteile einer solchen Behandlung müssen allerdings gegen die potenziellen Risiken abgewogen werden: eine mögliche Verlängerung der QT-Zeit, die häufig Auslöser maligner Herzrhythmusstörungen ist, allergische Reaktionen und die Zunahme makrolidresistenter Stämme von S. pneumoniae und H. influenzae, mit der zu rechnen ist und die aus epidemiologischer Sicht ungünstig ist.
Statine haben neben ihrer lipidsenkenden auch antioxidative und antiinflammatorische Wirkungen. Die Konzentrationen von IL-6, TNF alpha sowie das high sensitivity(hs)-CRP werden gesenkt. hsCRP ist ein Marker der systemischen Inflammation, der bei COPD selbst in einer stabilen Krankheitsphase erhöht ist. (30). Unter Statinen wurde in Metaanalysen nicht nur eine Senkung der kardiovaskulären, sondern auch der respiratorischen Letalität (um 18%) gefunden (31). Im Tierversuch wurde durch Statine Emphysembildung und pulmonale Hypertonie verzögert. Von einer generellen Empfehlung für Statine bei COPD kann jedoch keine Rede sein.
Neue Arten der Therapie sind dringend nötig, denn mit keiner etablierten Therapie kann bisher die Letalität bei COPD gesenkt werden. Die Tabakentwöhnung ist zwar effektiv und kostengünstig, wird aber trotz guter Datenlage nur unzureichend propagiert und erreicht. Sie ist bisher die einzige Maßnahme, die die kontinuierliche Verschlechterung des FEV1 aufhalten kann (32). Eine Reduktion der Krankheitsprogression um 50% ist bei strikter Tabakkarenz möglich (33, 34). Dies ist mit keinem Arzneimittel bisher zu erreichen!
Literatur
- Murray, C.J., und Lopez, A.D.: Lancet 1997, 349, 1498. Link zur Quelle
- König, G.: Atemw.-Lungenkrkh. 2004, 30, 181.
- Profita, M., et al.: Allergy 2005, 60, 1361. Link zur Quelle
- Løkke, A., et al.: Thorax 2006, 61, 935. Link zur Quelle
- Lange, P., et al. (CCHS =
- www.copd.versorgungsleitlinie.de Link zur Quelle
- Vogelmeier, C., et al.: Leitlinie der deutschen Atemwegsliga. Pneumologie 2007, 61, e1 Link zur Quelle. Erratum: Pneumologie 2007, 61, 517.
- Celli, B.R., et al.: N. Engl. J. Med. 2004, 350, 1005. Link zur Quelle
- Fletcher, C., und Peto, R.: Brit. Med. J. 1977, 1, 1645. Link zur Quelle
- Hamm, H.: Internist 2006, 47, 901. Link zur Quelle
- AMB 2003, 37, 09. Link zur Quelle
- Casaburi, R., und ZuWallak, R.: N. Engl. J. Med. 2009, 360, 1329. Link zur Quelle
- van Noord, J.A., et al.: Chest 2006, 129, 509. Link zur Quelle
- Tashkin, D.P., et al. (UPLIFT = Understanding Potential Long-term Impacts on Function with Tiotropium): N. Engl. J. Med. 2008, 359, 1543. Link zur Quelle
- Lee, T.A., et al.: Ann. Intern. Med. 2008, 149, 380. Link zur Quelle
- Calverley, P.M., et al. (TORCH = TOwards a Revolution in COPD Health): N. Engl. J. Med. 2007, 356, 775. Link zur Quelle
- Drummond, H.B., et al.: JAMA 2008, 300, 2407. Link zur Quelle
- AMB 2003, 37, 17. Link zur Quelle
- Dtsch. Arztebl. 2008, 105, 424. Link zur Quelle
- Decramer, M., et al. (BRONCUS = Bronchitis Randomized On NAC Cost-Utility Study): Lancet 2005, 365, 1552 Link zur Quelle. Erratum 2005, 366, 984.
- Decramer, M., et al.: Thorax 2005, 60, 343. Link zur Quelle
- AMB 2005, 39, 52. Link zur Quelle
- Barnes, P.J., und Stockley, R.A.: Eur. Respir. J. 2005, 25, 1084. Link zur Quelle
- Sethi, S., und Murphy, T.F.: N. Engl. J. Med: 2008, 359, 2355. Link zur Quelle
- Mahler, D.A., et al.: Chest 2004, 126, 926. Link zur Quelle
- Compton, C.H., et al. (ISG = International Study Group): Lancet 2001, 358, 265. Link zur Quelle
- AMB 2005, 39, 69b. Link zur Quelle
- Suzuki, T., et al.: Chest 2001, 120, 730. Link zur Quelle
- Seemungal, T.A., et al.: Am. J. Respir. Crit. Care Med. 2008, 178, 1139 18723437. Link zur Quelle
- Watz, H., und Magnussen, H.: Internist 2006, 47, 895. Link zur Quelle
- Baigent, C., et al. (CCT = Cholesterol Treatment Trialists‘ Collaborators): Lancet 2005, 366, 1267 Link zur Quelle. Errata: Lancet 2005, 366, 1358 und Lancet 2008, 371, 2084.
- Simmons, M.S., et al. (LHS = Lung Health Study): Eur. Respir. J. 2005, 25, 1011. Link zur Quelle
- Barnes, P.J.: Pharmacol. Rev. 2004, 56, 515. Link zur Quelle
- Donohue, J.F.: Lancet 2005, 365, 1518. Link zur Quelle