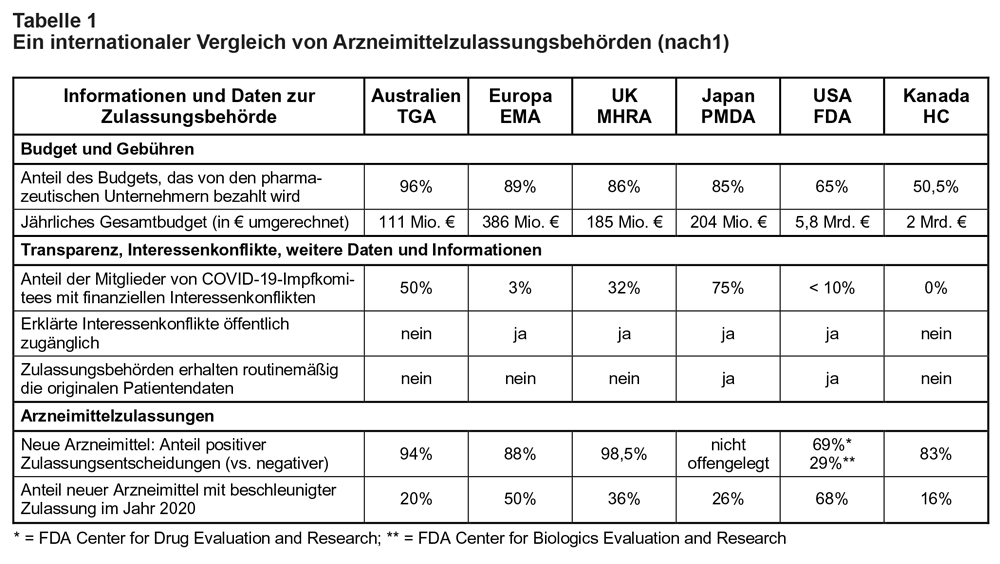
Eine investigative Journalistin, Maryanne Demasi, hat für das „British Medical Journal“ (BMJ) eine Untersuchung durchgeführt, die sich mit der Frage der Unabhängigkeit von sechs weltweit führenden Zulassungsbehörden für Arzneimittel von pharmazeutischen Unternehmern (pU) beschäftigt ([1]). Das BMJ befragte zu diesem Zweck folgende sechs Arzneimittelzulassungsbehörden: „Therapeutic Goods Administration“ (TGA; Australien); „European Medicines Agency“ (EMA, Europa); „Medicines and Healthcare Products Regulatory Agency“ (MHRA, Großbritannien); „Pharmaceuticals and Medical Devices Agency“ (PMDA, Japan), „US Food and Drug Administration“ (FDA, USA); „Health Canada“ (HC, Kanada). Die an diese Zulassungsbehörden gerichteten Fragen bezogen sich auf ihre Finanzierung, Transparenz der im Rahmen der Zulassung ausgewerteten Daten, maßgebende Gründe für ihre Entscheidungen und auf die Geschwindigkeit, mit der neue Arzneimittel bewertet und zugelassen werden. Darüber hinaus wurden 4 renommierte Experten (Mediziner und Soziologen) befragt, die sich seit vielen Jahren wissenschaftlich mit der Frage der heute häufig beschleunigten Zulassungsverfahren von Arzneimitteln sowie der Beeinflussung der Zulassungsverfahren und deren Entscheidungen durch finanzielle Interessenkonflikte beschäftigt haben (Courtney Davis, Aaron Kesselheim, Joel Lexchin, Donald Light). In einer sehr informativen Tabelle werden die sechs o.g. Zulassungsbehörden verglichen, u.a. hinsichtlich ihres jährlichen Budgets bzw. des Prozentsatzes des von pU beigesteuerten Budgets, der Transparenz in Bezug auf die Deklaration von Interessenkonflikten der beteiligten Experten sowie hinsichtlich der Datensätze der in den klinischen Studien rekrutierten Patienten. Außerdem wurde der Anteil ermittelt der zugelassenen neuen Arzneimittel bezogen auf alle beantragten Zulassungen bzw. der Prozentsatz an beschleunigten Zulassungen im Jahr 2020 (vgl. Tab. [1]).
Die Ergebnisse sind ernüchternd, beispielsweise in Hinsicht auf die Finanzierung der in den USA im Rahmen des 1992 verabschiedeten „Prescription Drug User Fee Act“ (PDUFA) bewerteten Zulassungsanträge und ihren Auswirkungen. Die zunehmende Akzeptanz industrieller Gebühren im Rahmen der Zulassung hat in den USA und auch in Australien dazu geführt, dass die Anforderungen an die in klinischen Zulassungsstudien erbrachte Evidenz für Wirksamkeit und Sicherheit neuer Arzneimittel abgenommen haben und dadurch vermutlich Patienten Schaden zugefügt wurde.
Der US-amerikanische Soziologe Donald Light kritisiert dieses Vorgehen, da die überwiegende Finanzierung „unabhängiger Institutionen“ (wie beispielsweise TGA, EMA und FDA) durch Gebühren der pU, deren neue Arzneimittel beurteilt und ggf. zugelassen werden sollen, einen fundamentalen Interessenkonflikt darstellt und somit aus seiner Sicht ein Paradebeispiel für institutionelle Korruption veranschaulicht ([1]). Er fordert deshalb Ärzte und Patienten auf, selber einzuschätzen, inwieweit Entscheidungen der Zulassungsbehörden mit überwiegend industrieller Finanzierung noch vertrauenswürdig sind. Die Besorgnis über Auswirkungen finanzieller Interessenkonflikte beschränkt sich in diesem Zusammenhang keineswegs nur auf die Mitarbeiter der Zulassungsbehörden, sondern vor allem auch auf die externen Mitglieder der Gremien, die in beratender Funktion an Zulassungsentscheidungen, derzeit besonders häufig auch für Impfstoffe gegen SARS-CoV-2, beteiligt sind ([2], [3]). Wie berechtigt diese Besorgnis ist, wird durch zahlreiche Untersuchungen bestätigt, so auch durch eine größere Studie, in der über einen Zeitraum von 15 Jahren die Auswirkungen von finanziellen Interessenkonflikten der Mitglieder in FDA-Beratungsgremien untersucht wurden ([4]). Auch diese Untersuchung ergab, dass Mitglieder mit Interessenkonflikten häufiger eine positive Bewertung des gesponserten Produkts abgaben.
Kritisiert wird in dem aktuellen BMJ Artikel auch das sog. Drehtürprinzip („revolving door“): Früher für Zulassungsbehörden tätige Experten arbeiten oder beraten nach Beendigung dieser Tätigkeit für dieselben pU, deren Produkte sie zuvor beurteilt hatten ([1]). Verdeutlicht wird dies am Beispiel der FDA, in der 9 von 10 Kommissionsmitgliedern im Zeitraum von 2006 bis 2019 heute wieder für pU arbeiten bzw. sie beraten. Dies gilt auch für die Beratung von Politikern hinsichtlich des Nutzens von Impfstoffen und Medikamenten bei COVID-19 (2, 3). Für den Umgang mit diesen sogenannten „pay later“-Interessenkonflikten werden Regeln gefordert, wie beim Seitenwechsel von der Politik in die Wirtschaft beispielsweise eine Karenzzeit (sogenannte Abklingzeit; vgl. [5]).
In einem eigenen Abschnitt dieser Publikation werden notwendige strukturelle Reformen diskutiert, um die Fähigkeit der Zulassungsbehörden, unabhängige Entscheidungen zu treffen, wiederherzustellen – frei von Beeinflussung durch industrielle Interessen ([1]). Die befragten Experten schlagen hierfür verschiedene Maßnahmen vor. Zunächst muss die Transparenz hinsichtlich der finanziellen Interessenkonflikte bei Beratern der Zulassungsgremien deutlich verbessert und darüber hinaus auch begründet werden, weshalb Experten mit relevanten finanziellen Interessenkonflikten an Entscheidungsprozessen der Zulassungsbehörden beteiligt sind. Ein weiterer, auch aus unserer Sicht wichtiger Schritt ist die stärkere Überprüfung beschleunigter Zulassungsverfahren bei neuen Arzneimitteln, z.B. hinsichtlich der ausgewählten Endpunkte in klinischen Studien und deren Aussagekraft für die Beurteilung des Nutzens neuer Wirk-/Impfstoffe ([6]). Ein Experte (Donald Light) glaubt jedoch nicht mehr daran, dass Zulassungsbehörden künftig unvoreingenommene, präzise Bewertungen neuer Arzneimittel vornehmen werden und schlägt stattdessen vor, dass unabhängige „Non-Profit“-Organisationen“ wie bspw. „Health-Technology Assessment“ (HTA)-Institute diese außerordentlich wichtige, auf wissenschaftlicher Evidenz basierende Aufgabe übernehmen sollten ([1]).
Literatur
- Demasi, M.: BMJ 2022, 377, o1538
- Thacker, P.D.: BMJ 2020, 371, m4716
- Thacker, P.D.: BMJ 2021, 373, n1283
- Pham-Kanter, G.: Milbank Q. 2014, 92, 446.
- AMB 2018, 52, 72DB01 (Link zur Quelle)
- Ludwig, W.-D.: Zulassungsverfahren neuer Arzneimittel in Europa. In: Schwabe, U., Paffrath, D., Ludwig, W.-D., Klauber, J. (Hrsg.): Arzneiverordnungs-Report 2019. Springer-Verlag 2019