Über die Probleme bei Bewertung, Zulassung (pre-market approval) sowie Überwachung (post-market surveillance) verschiedener Medizintechnikprodukte haben wir mehrfach berichtet (1-6). Neben teilweise unvermeidbaren produktspezifischen Besonderheiten, z.B.: eine Randomisierung, Plazebokontrolle und/oder Verblindung häufig nicht möglich oder ethisch nicht vertretbar, rasche Obsoleszenz (Veralten) infolge technologischer Weiterentwicklung, gab insbesondere das europäische System der CE-Zertifizierung durch dezentrale sogenannte „Benannte Stellen“ („Notified Bodies“) immer wieder Anlass zur Kritik.
Im April 2017 wurden von der EU neue Verordnungen zu Medizintechnik-Produkten (MP-VO; englisch: Medical Device Regulation = MDR) und In-vitro-Diagnostika (IVD-VO; In-vitro Device Regulation = IVDR) erlassen (7), deren zeitlicher Beginn – nach Ablauf der jeweiligen Übergangsfristen – für den 26. Mai 2020 (MP-VO) bzw. für den 26. Mai 2022 (IVD-VO) festgesetzt wurden. Für die Hersteller von Medizinprodukten und die „Benannten Stellen“ resultieren aus dieser Novelle größere Änderungen. Zusätzlich zu den bereits in den jeweiligen vorausgegangenen Verordungen enthaltenen Anforderungen, von denen keine gestrichen wurde, kamen zahlreiche, deutlich strengere Anforderungen hinzu:
-
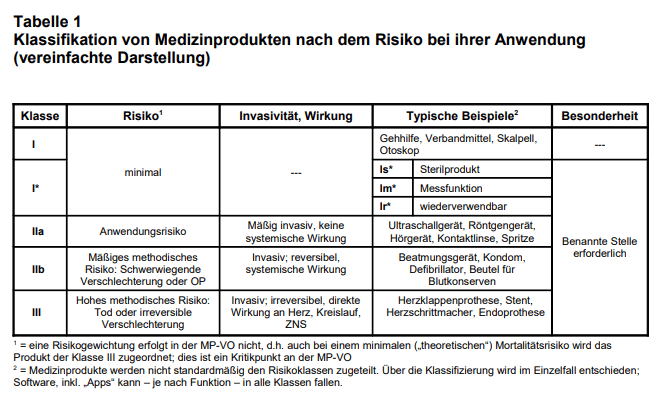
Strengere Klassifizierungsregeln: Die grundlegende Klassifizierung von Medizinprodukten bleibt im Wesentlichen unverändert (s. Tab. 1), aber es erfolgt eine Neueinstufung der Produkte nach strengeren Regeln. Einige Produkte (einschließlich Software-Applikationen) fallen dadurch in höhere Risikoklassen als bisher; einige Produkte wurden neu als Medizinprodukte eingestuft.
-
Strengere klinische Prüfung und Bewertung: Eine Zulassung anhand von Äquivalenzdaten (Vergleichsdaten bestehender Produkte) ist jetzt nur noch in Ausnahmefällen möglich. Dadurch müssen Hersteller klinische Studien in viel größerem Umfang als bisher durchführen; die Anforderungen an klinische Studien wurden erhöht.
-
Höhere Anforderungen an „Benannte Stellen“: Diese übernehmen neue Überwachungsfunktionen; fachliche Eignung und klinische Kompetenz erhalten einen höheren Stellenwert. Alle „Benannten Stellen“ müssen eine Neubenennung beantragen.
-
Erhöhte Transparenz durch Identifizierbarkeit und Rückverfolgbarkeit von Produkten: Zur eindeutigen Produktkennzeichnung wird eine spezifische Produktnummer eingeführt (Unique Device Identification = UDI); die UDI-Datenbank wird Bestandteil der erweiterten Europäischen Datenbank für Medizinprodukte (EUDAMED).
-
Höhere Anforderungen bei Vigilanz und Marktüberwachung: Unangekündigte Audits, Stichproben und Produktprüfungen nach Marktzulassung sind vorgesehen; bei definierten Produktgruppen müssen die Hersteller jährliche Berichte über Qualität, Sicherheit und Leistung über den gesamten Produktlebenszyklus einreichen. Jeder Hersteller muss eine dafür „Verantwortliche Person“ benennen.
-
Anspruchsvollere Konformitätsbewertungsverfahren: Die Beurteilung der Konformität eines Produkts mit den regulatorischen Anforderungen durch Hersteller und Benannte Stellen wird deutlich aufwändiger. Bei bestimmten Risikoprodukten ist die Konsultation eines unabhängigen Expertengremiums vorgeschrieben.
-
Kein Bestandschutz: Alle derzeit bereits genehmigten Medizinprodukte müssen nach den genannten neuen Anforderungen rezertifiziert werden. Über Ausnahmen wird verhandelt.
Über (fehlende) Konsistenz und Präzision in der praktischen Umsetzung dieser grundsätzlich begrüßenswerten Änderungen wird seit dem Erlass der Verordnung vor drei Jahren heftig debattiert. Fest steht, dass diese Anforderungen die Hersteller von Medizinprodukten unter beträchtlichen Druck setzen. Insbesondere die zeitaufwändigen Konformitätsbewertungen durch „Benannte Stellen“ infolge der Re-Klassifizierungen von hunderttausenden bereits zugelassenen Produkten sind eine Herausforderung, zumal – ebenfalls als Folge der neuen Bestimmungen – die Zahl der (Neu-)“Benannten Stellen“ im selben Zeitraum stark abgenommen hat. Da eine fristgerechte Umsetzung ganz offensichtlich nicht mehr möglich war, veröffentlichte die EU bereits im Dezember 2019 ein schon lange von Herstellern gefordertes Korrigendum, welches unter anderem vorsah, dass bestimmte Produkte noch bis Mai 2024 in Verkehr gebracht werden können (zugelassene Medizinprodukte der bisherigen Klasse I, die gemäß der alten Richtlinie genehmigt, aber nun höher zu klassifizieren und erstmals durch Benannte Stellen zu zertifizieren sind = Klasse I*; s. Tab. 1).
Als Folge der SARS-CoV-2-Pandemie wurde nun in einer aktuellen EU-Verordnung vom 23. April 2020 der Termin für den Geltungsbeginn der gesamten MP-VO um ein Jahr auf den 26. Mai 2021 verschoben (8). Diese Entscheidung sei aufgrund der unerwarteten, außergewöhnlich großen Nachfrage nach lebenswichtigen Medizinprodukten, z.B. persönliche Schutzausrüstung, Material für Intensivpflege, Respiratoren, erforderlich geworden. Die ordnungsgemäße Durchführung und Anwendung der MP-VO zum festgelegten Geltungsbeginn am 26. Mai 2020 sei in dieser Situation nicht sichergestellt. Außerdem müsse man weitere Materialengpässe und Verzögerungen infolge gebundener behördlicher Kapazitäten vermeiden. Der Geltungsbeginn der IVD-VO am 26. Mai 2022 ist von dieser aktuellen Terminverschiebung nicht betroffen.
Neben der Terminverschiebung um ein Jahr wurde mit derselben Verordnung auch eine Ausweitung von in der MP-VO vorgesehenen und genehmigten nationalen Ausnahmeregelungen (erleichterte Produktzulassung „im Interesse der öffentlichen Gesundheit oder der Patientensicherheit oder -gesundheit“) auf das gesamte Gebiet der EU ermöglicht (8). Somit ist damit zu rechnen, dass (vorübergehend) Medizinprodukte zur medizinischen Anwendung am Menschen kommen, die keine Konformitätserklärung bzw. keine CE-Kennzeichnung haben. Dies wird aller Voraussicht nach insbesondere bei Produkten der Fall sein, die in der Versorgung von COVID-19-Patienten eingesetzt werden. Die Gewährleistung, dass es sich hier nicht um eine „neue Normalität“, sondern um einen vorübergehenden Ausnahmezustand handelt, sollte aus unserer Sicht hohe Priorität haben, um zu verhindern, dass Sicherheitsstandards aufgeweicht werden, noch bevor sie überhaupt in Kraft treten.
Fazit: Am 26. Mai 2020 sollte eine wichtige und aus unserer Sicht lange überfällige Novelle der europäischen Medizinprodukte-Verordnung in Kraft treten. Um eine Überlastung behördlicher Kapazitäten und damit Versorgungsengpässe bei lebenswichtigen Medizinprodukten während der SARS-CoV-2-Pandemie zu vermeiden, wurde dieser Termin nun nach einem EU-Beschluss um ein Jahr verschoben. Außerdem wurde die Möglichkeit geschaffen, erleichterte Produktzulassungen, die als nationale Ausnahmeregelungen vorgesehen waren, nun EU-weit zu genehmigen. Diese Entscheidung kommt in Anbetracht der derzeitigen Unwägbarkeiten vielen Beteiligten entgegen – insbesondere wohl auch den Herstellern von Medizinprodukten. Eine Rückkehr zur Umsetzung der neuen Medizinprodukte-Verordnung in der ursprünglich geplanten Form und zu einem frühen möglichen Termin halten wir dringend erforderlich.
Literatur
- Allgemein: AMB 2017, 51, 31. Link zur Quelle AMB 2014, 48, 30b. Link zur Quelle
- Herzschrittmacher: AMB 2020, 54, 24DB01. Link zur Quelle
- Herzklappen: AMB 2012, 46, 89. Link zur Quelle
- Kardiovaskuläre Implantate: AMB 2010, 44, 09. Link zur Quelle
- Brustimplantate: AMB 2012, 46, 15a. Link zur Quelle
- Hüftendoprothesen: AMB 2012, 46,88DB01. Link zur Quelle
- https://eur-lex.europa.eu/… Link zur Quelle
- https://eur-lex.europa.eu/… Link zur Quelle