Gesetz zur Neuordnung des Arzneimittelmarktes: harmonischer Dreiklang oder eintönige Preisregulierung?
Die Arzneimittelausgaben der gesetzlichen Krankenversicherung (GKV) sind in den letzten Jahren in Deutschland weiter stark gestiegen und liegen seit dem Jahr 2001 über den Ausgaben für ärztliche Behandlung (1). Verursacht wird der Anstieg in erster Linie durch höhere Ausgaben für Arzneimittel ohne Festbetrag (Anstieg 2009 um 8,9%) sowie kostenintensive Spezialpräparate mit jährlich zweistelligen Wachstumsraten (1, 2, 23). Aus Sicht des Bundesministeriums für Gesundheit (BMG) besteht nun – auch unter dem zunehmenden Kostendruck im Gesundheitswesen – kurzfristiger Handlungsbedarf, dem die Fraktionen der CDU/CSU und FDP mit dem Gesetz zur Neuordnung des Arzneimittelmarktes in der GKV (AMNOG) nachgekommen sind (3). Laut einer Pressemitteilung des BMG beinhaltet das am 11. November 2010 verabschiedete AMNOG einen „Dreiklang aus strukturellen Veränderungen, dem Abbau von Überregulierung und kurzfristigen Einsparungen“ (4). Während fast alle anderen Länder in der Europäischen Union (EU) bereits seit einigen Jahren rasch nach der Zulassung in Verhandlungen mit dem pharmazeutischen Unternehmer (PU) einen am Nutzen orientierten Preis festsetzen, besteht in Deutschland bis heute die Möglichkeit der freien Preisbildung für patentgeschützte Arzneimittel und gleichzeitig dient Deutschland als Referenzland für Arzneimittelpreise in mehreren europäischen Staaten (1, 5, 24).
Das AMNOG erwartet – zusammen mit einer bereits am 30. Juli 2010 in Kraft getretenen Regelung im Gesetz zur Änderung krankenversicherungsrechtlicher und anderer Vorschriften (GKV-ÄndG) – Einsparungen in Höhe von 2,4 Mrd. € pro Jahr, davon 2,2 Mrd. € für die GKV (4). Die kurzfristig angestrebten Einsparungen bei den Arzneimittelkosten ab dem Jahr 2011 umfassen verschiedene Maßnahmen. Hierzu zählen u.a. die Erhöhung der gesetzlichen Hersteller-Rabatte für Arzneimittel ohne Festbetrag von 6% auf 16% vom 1. August 2010 bis zum 31. Dezember 2013 einschließlich Preismoratorium, die Anhebung des Apothekenrabatts von 1,75 € auf 2,05 € je Packung verschreibungspflichtiger Arzneimittel in den Jahren 2011 und 2012 (nur GKV), eine Senkung des Volumens des Großhandelzuschlags für rezeptpflichtige Arzneimittel (GKV und private Krankenversicherung = PKV) und die Übertragung der gesetzlichen Hersteller-Rabatte auf die PKV (3).
Im Mittelpunkt dieses Artikels stehen die als „strukturelle, langfristig wirksame Veränderungen“ apostrophierten Reformen im AMNOG, die zu Änderungen des § 35a, § 92, § 130 des Sozialgesetzbuchs (SGB) Fünftes Buch (V) und einer Änderung des § 42b im Arzneimittelgesetz (AMG) führen.
Was wissen wir über neue Arzneimittel zum Zeitpunkt der Zulassung? Im Rahmen der Zulassung werden Qualität, Wirksamkeit und Sicherheit eines neuen Wirkstoffs in dem vorgesehenen Anwendungsgebiet von den nationalen Zulassungsbehörden bzw. der European Medicines Agency (EMA) geprüft und bei Vorliegen eines positiven Nutzen-Risiko-Verhältnisses („Benefit-risk-profile“) die Zulassung erteilt (6, 7). Pharmakoökonomische Aspekte spielen bei den Zulassungsentscheidungen keine Rolle. Dabei sollten – entsprechend der Richtlinie 2001/83/EG zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel – klinische Prüfungen in der Regel als „kontrollierte klinische Prüfungen“ und soweit möglich randomisiert durchgeführt werden, wobei zum Vergleich je nach Einzelfall ein Plazebo oder ein bereits bekanntes Arzneimittel mit nachgewiesenem therapeutischen Wert heranzuziehen ist (6). Untersuchungen in den letzten Jahren haben jedoch gezeigt, dass diese Vorgaben von der EMA nicht immer konsequent eingehalten wurden. So wurde nur in etwa 50% der in den europäischen Bewertungsberichten (European Public Assessment Report = EPAR) erwähnten klinischen Studien (Zeitraum der Auswertung: 1. Januar 2007 bis 31. Dezember 2008) der neue Wirkstoff mit einem Arzneimittel mit nachgewiesener therapeutischer Wirksamkeit („active comparator“) verglichen und nur in etwa 20% war das Design der Zulassungsstudie darauf ausgerichtet, eine Überlegenheit des neuen Wirkstoffs im Vergleich zum „active comparator“ in randomisierten kontrollierten Studien (RCT) zu belegen (7). Dies hat zur Folge, dass in Zulassungsstudien beim Vergleich neuer mit bereits vorhandenen Arzneimitteln sehr häufig eine „Gleichwertigkeit“ („Equivalence“) oder sogar eine „nicht nachgewiesene Unterlegenheit“ („Non-inferiority“) des neuen Wirkstoffs als für die Wirksamkeit ausreichender Beleg akzeptiert wird (8, 9). Weitere Merkmale, die die Übertragbarkeit der Ergebnisse aus Zulassungsstudien zu neuen Arzneimitteln auf die Behandlung von Patienten unter Alltagsbedingungen in Klinik oder Praxis (externe Validität) einschränken, sind die häufig strikten Ein- und Ausschlusskriterien, Verwendung von Surrogat- bzw. kombinierten „Outcome“-Parametern als primäre Endpunkte und die in Zulassungsstudien häufig kurzen Zeiträume der Behandlung bzw. Nachbeobachtung (10, 11). Endgültige Aussagen zur Sicherheit bzw. zu den unerwünschten Arzneimittelwirkungen (UAW) neuer Wirkstoffe sind in Zulassungsstudien nur sehr eingeschränkt möglich, da aufgrund der kleinen Patientenzahlen und in der Regel kurzen Studiendauer bzw. Nachbeobachtung nur (sehr) häufige bzw. akut auftretende UAW erfasst werden. Bei der Zulassung neuer Wirkstoffe werden deshalb heute im Rahmen der Pharmakovigilanz weitere Untersuchungen zur Sicherheit gefordert.
Mit der Zulassung wird einem neuen Arzneimittel ein positives „Nutzen (besser Wirksamkeit)-Risiko-Verhältnis“ attestiert. Zu diesem Zeitpunkt stehen aber nur begrenzt Ergebnisse aus klinischen Studien zu Wirksamkeit sowie Sicherheit zur Verfügung. Vergleiche zur medikamentösen Standardtherapie bzw. zu nicht-medikamentösen Therapiestrategien, die für eine Bewertung des Nutzens bzw. Zusatznutzens des neuen Wirkstoffs benötigt werden, liegen selten vor (7-9, 12). Dies wird auch aus Sicht der PU nicht bestritten (13). Zielsetzungen von AMG und SGB V sind somit auch unterschiedlich. Anders als bei der Bewertung der Wirksamkeit eines neuen Arzneimittels unter den Bedingungen einer klinischen Zulassungsstudie, die eher den Idealfall als die Realität widerspiegeln, stehen bei der sozialrechtlichen Bewertung Aspekte der therapeutisch und wirtschaftlich effizienten Versorgung von Patienten im Vordergrund. Entsprechend § 35b im SGB V sind beim Nutzen für die Patienten insbesondere die Verbesserung des Gesundheitszustands, eine Verkürzung der Krankheitsdauer, eine Verlängerung der Lebensdauer, eine Verringerung der Nebenwirkungen sowie eine Verbesserung der Lebensqualität zu berücksichtigen. Dabei beruht die Nutzenbewertung auf einer vor der Zulassung nicht möglichen Abwägung zwischen positiven (Wirksamkeit) und negativen (Risiken bzw. Schäden) therapeutischen Effekten unter Alltagsbedingungen im Hinblick auf die Anwendungsgebiete des neuen Wirkstoffs. Sie sollte stets an Hand von Vergleichen mit den geltenden medizinischen Standards erfolgen (14).
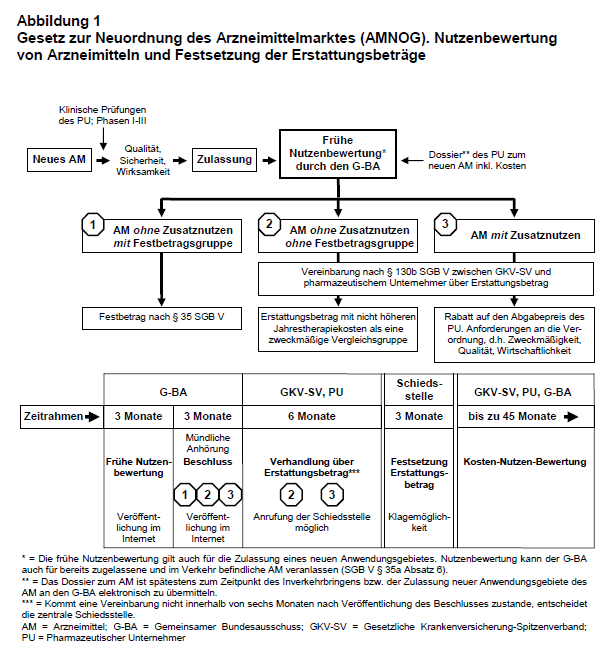
Frühe (Zusatz-)Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen: Im Mittelpunkt des AMNOG steht die sogenannte frühe Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen bzw. neuen Anwendungsgebieten, die vom Gemeinsamen Bundesausschuss (G-BA) vorgenommen wird. Der neue § 35a des SGB V sieht vor, dass zur Nutzenbewertung insbesondere die Bewertung des Zusatznutzens gegenüber der zweckmäßigen Vergleichstherapie sowie des Ausmaßes des Zusatznutzens und seiner therapeutischen Bedeutung gehört. Der Begriff des Zusatznutzens ist relativ neu im SGB V und gesetzlich nicht definiert (15). Verbraucherorganisationen in der EU sprechen von einem Zusatznutzen, wenn zuverlässige Ergebnisse klinischer Prüfungen vorliegen, die für Patienten eine bessere Wirksamkeit und/oder größere Sicherheit und/oder einfachere Verabreichung neuer Arzneimittel im Vergleich zu den vorhandenen Alternativen zeigen (7). Grundlage der Nutzenbewertung sind Nachweise des PU, die er als Dossier, einschließlich aller von ihm durchgeführten oder in Auftrag gegebenen klinischen Prüfungen, spätestens zum Zeitpunkt des erstmaligen Inverkehrbringens bzw. der Zulassung neuer Anwendungsgebiete des Arzneimittels an den G-BA elektronisch zu übermitteln hat. Die Angaben, die dieses Dossier enthalten muss, sind insbesondere: zugelassene Anwendungsgebiete, medizinischer Nutzen bzw. Zusatznutzen im Verhältnis zur zweckmäßigen Vergleichstherapie und Kosten der Therapie für die GKV.
Bei Arzneimitteln, die pharmakologisch-therapeutisch vergleichbar mit Festbetragsarzneimitteln sind, ist der medizinische Zusatznutzen, wie bisher im SGB V, als therapeutische Verbesserung entsprechend § 35 (Festbeträge für Arznei- und Verbandmittel) nachzuweisen. Für Arzneimittel mit neuen Wirkstoffen, die pharmakologisch-therapeutisch nicht vergleichbar sind mitFestbetragsarzneimitteln, erfolgt der Nachweis eines medizinischen Zusatznutzens im Vergleich mit einer nach dem allgemeinen Stand der medizinischen Erkenntnisse zweckmäßigen Therapie im Anwendungsgebiet. Grundsätzlich führt der G-BA die Nutzenbewertung auf der Basis des Dossiers des PU selbständig durch oder beauftragt das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) oder Dritte mit der Nutzenbewertung. Die Details der Nutzenbewertung wurden inzwischen in einem Entwurf einer Rechtsverordnung durch das BMG festgelegt, die in der endgültigen Form Anfang Januar 2011 in Kraft treten soll. Weitere Einzelheiten der Nutzenbewertung werden vom G-BA innerhalb eines Monats nach Inkrafttreten der Rechtsverordnung (d.h. voraussichtlich zum 31. Januar 2011) in einer Verfahrensordnung geregelt.
In der „frühen Nutzenbewertung“ (s. Abb. 1) wird für festbetragsfähige Arzneimittel mit neuen Wirkstoffen geprüft, ob durch sie eine therapeutische Verbesserung für Patientengruppen oder Anwendungsgebiete gegenüber der Festbetragsgruppe erreicht werden kann. Falls dies nicht der Fall ist, werden sie in die Festbetragsgruppe eingeordnet. Liegt eine therapeutische Verbesserung vor, wird entsprechend dem in das SGB V neu eingefügten § 130b zwischen dem GKV-Spitzenverband (GKV-SV) – im Benehmen mit dem Verband PKV – und dem PU ein Erstattungsbetrag vereinbart, der als Rabatt auf den Abgabepreis des PU gewährt wird. Bei nicht festbetragsfähigen Arzneimitteln mit neuen Wirkstoffen und gegenüber der zweckmäßigen Vergleichstherapie nachgewiesenem relevanten Zusatznutzen wird ebenfalls gemäß § 130b zwischen dem GKV-SV und dem PU ein Erstattungsbetrag vereinbart. Falls kein Zusatznutzen nachgewiesen und das Arzneimittel mit neuem Wirkstoff keiner Festbetragsgruppe zugeordnet werden kann, soll ein Erstattungsbetrag vereinbart werden, der nicht zu höheren Jahrestherapiekosten führt als die zweckmäßige Vergleichstherapie (§ 130b, Absatz 3). Falls zwischen GKV-SV und PU nicht innerhalb von sechs Monaten nach Veröffentlichung der Nutzenbewertung (d.h. 12 Monate nach Beginn der Bewertung des Nutzens von Arzneimitteln mit neuen Wirkstoffen) eine Vereinbarung zum Erstattungsbetrag zustande kommt, entscheidet eine zentrale Schiedsstelle innerhalb von drei Monaten über die Höhe des tatsächlichen Abgabepreises, der auch die Preise in anderen europäischen Ländern zu berücksichtigen hat.
Auch für bereits zugelassene und im Verkehr befindliche Arzneimittel kann der G-BA gemäß § 35a Absatz 6 des SGB V eine Bewertung veranlassen. Vorrangig sind Arzneimittel zu bewerten, die für die Versorgung von Bedeutung sind oder mit Arzneimitteln mit neuen Wirkstoffen im Wettbewerb stehen, für die ein Beschluss des G-BA hinsichtlich der Bewertung des Zusatznutzens vorliegt.
Arzneimittel für seltene Leiden: Im ursprünglichen Entwurf zum AMNOG war vorgesehen, dass für alle Arzneimittel mit neuen Wirkstoffen bzw. bei neuen Anwendungsgebieten bereits zugelassener Wirkstoffe ab 2011 eine frühe Nutzenbewertung erfolgen soll. In einem Änderungsantrag der Fraktionen von CDU/CSU und FDP wurde dann jedoch im § 35a Absatz 1 ein Satz angefügt, der besagt, dass bei Arzneimitteln zur Behandlung eines seltenen Leidens (Orphan Drugs = OD; vgl. 23) der medizinische Zusatznutzen durch die Zulassung belegt ist und Nachweise zum medizinischen Nutzen bzw. Zusatznutzen im Dossier des PU nicht vorgelegt werden müssen. Diese Änderung wurde damit begründet, dass bei Arzneimitteln für die Behandlung seltener Erkrankungen der im Rahmen der arzneimittelrechtlichen Zulassung erbrachte Wirksamkeitsnachweis grundsätzlich als Zusatznutzen im jeweiligen Anwendungsgebiet anzuerkennen ist und für die Behandlung dieser Erkrankung keine therapeutisch gleichwertige Alternative zur Verfügung steht. Diese Ausnahmeregelung für OD wurde scharf kritisiert (16) und im endgültigen Gesetzesentwurf wurde daraufhin im § 35a eine weitere Änderung eingefügt. Vom PU eines OD, dessen Umsatz in den letzten 12 Kalendermonaten einen Betrag von 50 Mio. € übersteigt, wird jetzt wieder der Nachweis des medizinischen Zusatznutzens gegenüber der zweckmäßigen Vergleichstherapie gefordert. Für OD mit einem jährlichen Umsatz ≤ 50 Mio. €/Jahr müssen entsprechende Nachweise jedoch nicht vorgelegt werden. Unabhängig vom Umsatz wird auch für alle neu zugelassenen OD ein Erstattungsbetrag zwischen GKV-SV und PU vereinbart.
Einschränkung oder Ausschluss der Verordnung von Arzneimitteln bei fehlendem Nachweis des therapeutischen Nutzens oder bei Unzweckmäßigkeit? Im § 92 des SGB V wurde bisher festgelegt, dass der G-BA über Richtlinien beschließen kann, die eine ausreichende, zweckmäßige und wirtschaftliche Versorgung der Versicherten garantieren. Dabei konnte er auch die Verordnung von Arzneimitteln einschränken oder ausschließen, wenn der therapeutische Nutzen, die medizinische Notwendigkeit oder die Wirtschaftlichkeit nicht nachgewiesen sind. In einem Änderungsantrag der Fraktionen CDU/CSU und der FDP vom 25.10.2010 zu Artikel 1 Nr. 13 (§ 92 SGB V) wurde die bisherige Formulierung im § 92 dahingehend verändert, dass der G-BA die Verordnung von Arzneimitteln nur einschränken oder ausschließen kann, wenn die Unzweckmäßigkeit erwiesen ist. Gleichzeitig wurde unter Punkt 2a ergänzt, dass der G-BA innerhalb einer angemessenen Frist ergänzende versorgungsrelevante Studien zur Bewertung der Zweckmäßigkeit eines Arzneimittels fordern kann. Das Nähere hierzu wird vom G-BA in seiner Verfahrensordnung geregelt.
Veröffentlichung der Ergebnisse klinischer Prüfungen:Das AMNOG beinhaltet auch eine Änderung des AMG. Im § 42b des AMG wird vom PU verlangt, der zuständigen Bundesoberbehörde Berichte über alle Ergebnisse konfirmatorischer klinischer Prüfungen zum Nachweis der Wirksamkeit und Unbedenklichkeit zur Eingabe in die Datenbank nach § 67a AMG innerhalb von sechs Monaten nach Zulassung (neu zugelassene Arzneimittel) bzw. innerhalb eines Jahres nach Beendigung der klinischen Studie (bereits zugelassene Arzneimittel) zur Verfügung zu stellen. Im Absatz 3 des § 42b wird ausdrücklich gefordert, dass die Ergebnisse der klinischen Prüfungen unabhängig davon, ob sie günstig oder ungünstig sind, in den Berichten enthalten sein müssen. Ferner werden Aussagen zu nachträglichen wesentlichen Prüfplanänderungen sowie Unterbrechungen und Abbrüchen der klinischen Prüfung in dem Bericht verlangt.
Was kann mit dem AMNOG erreicht werden und welche weiteren Schritte sind erforderlich auf dem Weg zu einer optimierten Einführung neuer Arzneimittel in die Versorgung?Die im AMNOG vorgesehene frühe Nutzenbewertung und die darauf basierende Vereinbarung eines Erstattungsbetrags zwischen GKV-SV und PU sind ein erster wichtiger Schritt auf dem Weg zu einer optimierten Markteinführung neuer Arzneimittel in Deutschland und zur Preisregulierung patentgeschützter Arzneimittel, die keiner Festbetragsgruppe zugeordnet werden können. Dieses Gesetz soll ab Anfang 2011 in Deutschland umsetzen, was in mehreren europäischen Ländern bereits seit Jahren, wenn auch unter unterschiedlichen nationalen Bedingungen, im Rahmen der Entscheidung über die Erstattungsfähigkeit eines neuen Arzneimittels und Festsetzung des Preises praktiziert wird (17, 18). Dabei wird auch in Deutschland die frühe Nutzenbewertung zwangsläufig aus einem anderen Blickwinkel (z.B. Bewertung des Zusatznutzens gegenüber der zweckmäßigen Vergleichstherapie) als dem der nationalen Zulassungsbehörden bzw. der EMA erfolgen. Die frühe Nutzenbewertung sollte jedoch nicht nur zur Preisregulierung und Kostenkontrolle genutzt werden, sondern auch einen Beitrag leisten, die Qualität der Arzneimitteltherapie zu verbessern. So ermöglicht z.B. die nach drei bzw. sechs Monaten im AMNOG vorgesehene Veröffentlichung der frühen Nutzenbewertung eine unabhängige Information von Ärzten und Apothekern, aber auch der Öffentlichkeit, über die verfügbaren wissenschaftlichen Erkenntnisse zu neuen Wirkstoffen bzw. neuen Anwendungsgebieten bereits zugelassener Wirkstoffe. Gleichzeitig kann besser als bisher auf die nur begrenzt zur Verfügung stehenden Daten zur Wirksamkeit und Sicherheit sowie den häufig umstrittenen oder nicht nachgewiesenen Zusatznutzen neuer Wirkstoffe im Vergleich zu medikamentösen Therapiealternativen hingewiesen werden. Von den Berichten des G-BA werden sicherlich auch unabhängige Arzneimittelinformationsblätter und die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) profitieren, deren wesentliches Ziel es ist, den rationalen Einsatz neuer Wirkstoffe zu propagieren und über deren Risiken rasch nach Zulassung zu informieren. Ein weiterer positiver Aspekt der Preisvereinbarungen auf Basis der frühen Nutzenbewertung könnte sein, dass die PU ihre derzeitige, eher an kommerziellen als an Patienteninteressen orientierte Vorgehensweise (z.B. neue Wirkstoffe schnell und zu hohen Preisen auf den Markt bringen) im Rahmen der Zulassungsstudien überdenken müssen. Die heute z.B. für Spezialpräparate häufig von den PU verlangten sehr hohen Preise sollten in Zukunft nur dann akzeptiert werden, wenn der (Zusatz-)Nutzen neuer Wirkstoffe gegenüber bereits verfügbaren Arzneimitteln durch Studien mit adäquatem Design belegt werden konnte, z.B. keine plazebokontrollierten Studien bei vorhandenen Therapiealternativen, Verzicht auf nicht-patientenrelevante Surrogat-Endpunkte, Nachweis der Überlegenheit statt Äquivalenz oder nicht-nachgewiesenen Unterlegenheit neuer Wirkstoffe (9, 11). Darüber hinaus werden die PU ihre Anstrengungen verstärken müssen, um an Hand validierter Parameter (z.B. Biomarker) Patientengruppen früher zu identifizieren, die von den neuen Wirkstoffen tatsächlich profitieren.
Verschiedene, kritisch zu bewertende Punkte im jetzt verabschiedeten Gesetz wurden von uns bereits dargestellt (2). Hierzu zählen insbesondere die nicht beabsichtigte Einführung eines Horizon Scanning Systems (HSS) in Deutschland, die weiterhin mögliche Vermarktung neuer Arzneimittel im ersten Jahr nach Zulassung mit vom PU selbst bestimmten, häufig überhöhten Preisen und die nach Festsetzung des Erstattungsbetrags nicht vorgesehenen Preis-Volumen-Absprachen mit gegebenenfalls Rückzahlungen bei Überschreiten der erwarteten Volumina. Ein nationales HSS für die frühe Identifizierung und Bewertung neuer Arzneimittel bzw. medikamentöser Therapiekonzepte wäre eine wichtige Ergänzung der frühen Nutzenbewertung gewesen, die jetzt vorwiegend auf dem Dossier des PU basiert. Die Möglichkeiten eines HSS, heute bereits in verschiedenen europäischen Ländern eingeführt und dort vorwiegend bei Health Technology Assessment (HTA)-Organisationen angesiedelt, sind an anderer Stelle ausführlich dargestellt (12).
Kritisiert werden muss am AMNOG aber auch, dass über die zahlreichen, z.T. offensichtlich auch unter dem Einfluss der PU formulierten Änderungsanträge zum ursprünglichen Gesetzesentwurf unter großem Zeitdruck und nicht immer mit adäquater Einbindung der Experten entschieden wurde. Dadurch wurden Regelungen aufgenommen (z.B. keine generelle frühe Nutzenbewertung für Arzneimittel gegen seltene Leiden, Umkehr der Beweislast und Nachweis der Unzweckmäßigkeit von Arzneimitteln durch den G-BA), die den ursprünglichen Zielen des AMNOG zuwiderlaufen. Damit diese, im AMNOG eingangs formulierten Ziele („den Menschen müssen im Krankheitsfall die besten und wirksamsten Arzneimittel zur Verfügung stehen sowie die Preise und Verordnungen von Arzneimitteln müssen wirtschaftlich und kosteneffizient sein“) tatsächlich erreicht werden, bedarf es neben der konsequenten Umsetzung der frühen Nutzenbewertung und Vereinbarung von Erstattungsbeträgen für alle neuen Arzneimittel, die keiner Festbetragsgruppe zugeordnet werden können, jedoch weiterer Maßnahmen. Sie betreffen sowohl die Anforderungen an die Zulassung von Arzneimitteln mit neuen Wirkstoffen als auch die rasche Durchführung unabhängiger klinischer Studien nach der Zulassung. Potenzielle Änderungen hinsichtlich Entwicklung und Zulassung neuer Wirkstoffe wurden kürzlich in einem Übersichtsartikel von Mitarbeitern der EMA ausführlich diskutiert (7).
Auf die für Patienten und Öffentlichkeit notwendigen Veränderungen bei der Zulassung und Bewertung neuer Arzneimittel in Europa haben italienische Pharmakologen hingewiesen (8, 9, 11, 19). Sie beziehen sich – neben dem Nachweis des Zusatznutzens neuer Wirkstoffe und der Forderung nach mehr unabhängigen klinischen Studien – insbesondere auf die stärkere Einbeziehung unabhängiger Patientengruppen bei Zulassungsentscheidungen sowie eine größere Transparenz der Rohdaten klinischer Zulassungsstudien, Studienprotokolle bzw. -ergebnisse sowie aller verfügbaren wissenschaftlichen Erkenntnisse, auf denen die Zulassung bzw. Verweigerung der Zulassung basiert. Durch die im AMG jetzt vorgeschriebene Veröffentlichung klinischer Prüfungen wird die Transparenz hinsichtlich der Studienergebnisse verbessert. Um Publikationsbias und die daraus resultierenden Nachteile bei der Nutzen-Risiko-Bewertung neuer Arzneimittel, systematischen Übersichtsarbeiten bzw. Metaanalysen und evidenzbasierten Therapieempfehlungen in Zukunft besser zu vermeiden (20-22), ist jedoch zu fordern, dass nicht nur alle Studienergebnisse und in Deutschland durchgeführten klinischen Studien mit Arzneimitteln, sondern auch das Studienprotokoll in einem zentralen, der Öffentlichkeit zugänglichen und vom PU unabhängigen Register eingesehen werden können.
Literatur
Schwabe, U., und Paffrath, D.: Arzneiverordnungs-Report 2010. Springer-Verlag Berlin Heidelberg New York, 2010.
Anders als in Europa sind in den USA die Zahlungen der Industrie an Ärztinnen und Ärzte transparent und bis zu jeder einzelnen Institution und Person in der „Open Payments Database“ nachvollziehbar . Im April haben wir darüber berichtet, dass von diesen Geldflüssen v.a. eine Minderheit profitiert und das negative Bild prägt (vgl. ). Etwa 6 von […]
Im JAMA ist im März ein „Research Letter“ erschienen, in dem die Zuwendungen (Geld und geldwerte Leistungen) der Industrie an Ärztinnen und Ärzte in den USA in den Jahren 2013-2022 untersucht wird . Die Informationen wurden aus der „Open Payments Platform“ extrahiert (vgl. ). Insgesamt wurden > 85 Mio. Transaktionen im Gesamtwert von 12,1 Mrd. US$ identifiziert. Pro Jahr wurden […]
Szenario: Eine deutsche Großstadt zu Beginn des Jahres 2024. Das lokale Krebszentrum veranstaltet in den eigenen Räumlichkeiten eine eintägige Konferenz, bei der kompakt über das Wichtigste und Neueste aus Hämatologie und Onkologie referiert wird. Jeder der 11 Themenblöcke wird in 2-4 „Impulsreferaten“ über 15 Minuten und einer 10-minütigen Diskussion abgehandelt. Die Referenten und Vorsitzenden kommen aus der […]
Die letzten Jahre waren finanziell sehr lukrative für die großen, global agierenden pharmazeutischen Unternehmer (pU). Nach einer Analyse von Macrotrends, einer Organisation, die für Investoren verschiedene Wirtschaftszweige an der Börse bewertet, haben die Top 15 der pU* im Jahr 2022 weltweit Umsätze von > 700 Mrd. US-$ getätigt und mit 146 Mrd. US-$ den höchsten Gewinn aller Zeiten eingestrichen. […]
Im Juli 2023 haben die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) und die Deutsche Gesellschaft für Allgemeinmedizin und Familienmedizin (DEGAM) eine gemeinsame Stellungnahme zur Einstellung von Produktion und Vertrieb sämtlicher Humaninsuline durch den pharmazeutischen Unternehmer Sanofi veröffentlicht . Wir schließen uns der darin geäußerten Kritik an und drucken die Stellungnahme hier mit kleinen Änderungen ab: Mit […]
Bereits Im November 2020 hatte die Europäische Kommission (EC) eine Arzneimittelstrategie für Europa vorgelegt, „mit der ein „zukunftssicherer und patientenorientierter Arzneimittelmarkt geschaffen werden sollte, auf dem die pharmazeutische Industrie in der Europäischen Union (EU) innovieren, florieren und weltweit führend bleiben kann“ . Diese Strategie stützte sich auf 4 Säulen: Gewährleistung des Zugangs für Patientinnen und Patienten […]
Die Entdeckung des Insulins, für die Frederick Grant Banting und John James Rickard Macleod im Jahr 1923 den Nobelpreis erhielten, ermöglichte die Entwicklung der ersten wirksamen Behandlung des Diabetes mellitus, der vorher innerhalb kurzer Zeit zum Tod geführt hatte . „Insulin gehört nicht mir, es gehört der Welt“ – mit diesen Worten verkaufte Banting mit […]
Die Geldflüsse zwischen der Industrie und medizinischen Dienstleistern sind auch 8 Jahre nach Etablierung des sog. „Transparenzkodex“ des Vereins „Freiwillige Selbstkontrolle für die Arzneimittelindustrie e.V.“ (FSA, vgl. ) sehr intransparent. Nach wie vor gibt es keine gemeinsame Darstellung der pharmazeutischen Unternehmer (pU) über ihre Zahlungen. Jeder pU veröffentlicht die jährlichen Zuwendungen an die Angehörigen der […]
Eine investigative Journalistin, Maryanne Demasi, hat für das „British Medical Journal“ (BMJ) eine Untersuchung durchgeführt, die sich mit der Frage der Unabhängigkeit von sechs weltweit führenden Zulassungsbehörden für Arzneimittel von pharmazeutischen Unternehmern (pU) beschäftigt (). Das BMJ befragte zu diesem Zweck folgende sechs Arzneimittelzulassungsbehörden: „Therapeutic Goods Administration“ (TGA; Australien); „European Medicines Agency“ (EMA, Europa); „Medicines […]
Etwa 50-65% der erwachsenen Patienten in den Hausarztpraxen klagen über Schlafstörungen. Dabei handelt es sich um ein breites Spektrum und bei weitem nicht nur um Ein- und Durchschlafstörungen. Die dritte Ausgabe der Internationalen Klassifikation von Schlafstörungen (ICSD-3-TR; ) nennt sieben Hauptkategorien (s. Tab. 1). Die exakte Diagnose ist entsprechend nicht einfach zu stellen. Oft überlappen sich die Kategorien […]