In den aktuell publizierten US-amerikanischen Leitlinien zur Behandlung von Vorhofflimmern (VHF) werden die direkten oralen Antikoagulanzien (DOAK) gegenüber den Vitamin-K-Antagonisten (VKA) bevorzugt empfohlen, wenn bei einem Patienten mit nicht-valvulärem VHF die Indikation zur oralen Dauerantikoagulation besteht (vgl. 1, 2). Es ist davon auszugehen, dass der bereits große Anteil von Patienten unter DOAK-Therapie weiter zunehmen wird. Ein gravierender Nachteil der DOAK ist das Fehlen eines spezifischen Antidots gegen direkte Faktor-Xa-Antagonisten (Rivaroxaban = Xarelto®, Apixaban = Eliquis®, Edoxaban = Lixiana®; vgl. 3). In Notfällen wird die Substitution von Gerinnungsfaktoren (Prothrombinkomplex) empfohlen. Lediglich für den direkten Thrombininhibitor Dabigatran liegt mit Idarucizumab ein seit 2015 in der EU und den USA zugelassenes Antidot vor (3). In den USA wurde im Mai 2018 mit Andexanet (Ondexxya®, Portola Pharmaceuticals) ein spezifisches Antidot gegen die Faktor-Xa-Antagonisten Apixaban und Rivaroxaban zugelassen mit der Indikation „lebensbedrohliche oder unkontrollierbare Blutungen“. Die EU-Zulassung steht unmittelbar bevor (s.u.). Es handelt sich um einen rekombinanten inaktiven Faktor Xa, der kompetitiv an die Faktor-Xa-Inhibitoren bindet und diese damit (partiell) inaktivieren soll. Die eingeschränkte und an Auflagen geknüpfte Zulassung erfolgte im Rahmen des „Accelerated Approval Program“ der FDA aufgrund zweier Studien zur Anti-Faktor-Xa-Senkung an gesunden Probanden (4) sowie vorab publizierter Daten der ANNEXA-4-Studie. In den oben erwähnten US-VHF-Leitlinien erhielt Andexanet eine Klasse-IIa-Empfehlung (Anm.: Idarucizumab: Klasse-I-Empfehlung; beides Evidenzlevel B). Die vollständigen Ergebnisse der ANNEXA-4-Studie, in der Andexanet erstmals an Patienten untersucht wurde, wurden jetzt im N. Engl. Med. publiziert (5). Die Studie wurde vom pharmazeutischen Unternehmer durchgeführt.
Methodik: Es handelt sich um eine nicht randomisierte, nicht kontrollierte, unverblindete („open label“) Kohortenstudie an Patienten mit akuten schweren Blutungen (hämodynamisch relevant oder Hb-Abfall ≥ 2 g/dl oder kritische Lokalisation, z.B. intrakraniell, retroperitoneal) innerhalb von 18 h nach Einnahme der Faktor-Xa-Inhibitoren Rivaroxaban, Apixaban, Edoxaban bzw. Injektion des niedermolekularen Heparins Enoxaparin (Clexane®) ≥ 1 mg/kg KG. Alle Patienten erhielten Andexanet als Bolus (400 mg oder 800 mg), gefolgt von einer Infusion (4 mg/min oder 8 mg/min) über 2 h. Die Dosierung war abhängig von Art, Zeitpunkt der letzten Einnahme und Dosis des Gerinnungshemmers mit Änderungen im Verlauf der Studie. Als primärer Endpunkt für die Wirksamkeit war initial allein das Erreichen einer prädefinierten Stillung der Blutung 24 h (später auf 12 h geändert) nach Ende der Infusion vorgesehen. Die Hämostase wurde durch eine unabhängige Kommission nach komplexen prädefinierten Kriterien in „sehr gut“, „gut“ oder „schlecht/keine“ klassifiziert (im „Supplementary Appendix“ nachzulesen; klinische und/oder Bildgebungskriterien waren abhängig von der Blutungslokalisation). Die Evaluierung, ob die Blutung gestillt war, erwies sich offenbar im Studienverlauf als problematisch, denn in einem späteren „Protocol Amendment“ wurde die gemessene Aktivität des Faktors Xa von einem initial sekundären zu einem zweiten „primären“ Endpunkt aufgewertet. Die beiden Endpunkte wurden sequenziell evaluiert, d.h. die Stillung der Blutung wurde nur dann evaluiert, wenn zuvor eine Änderung der Faktor-Xa-Aktivität nachgewiesen war. Außerdem erfolgte eine Analyse der Effektivität nur bei Patienten mit nachgewiesener schwerer Blutung und einer Anti-Faktor-Xa-Aktivität ≥ 75 ng/ml (bzw. ≥ 0,25 IU/ml nach Enoxaparin). Primäre Sicherheitsendpunkte waren Tod, thrombotische Ereignisse sowie die Bildung von Antikörpern gegen Andexanet, Faktor X oder Faktor Xa.
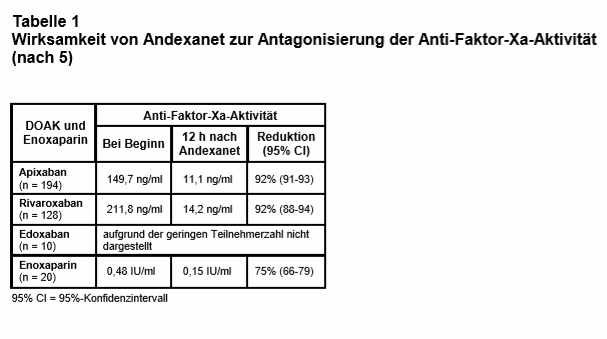
Ergebnisse: Es wurden zwischen April 2015 und Juni 2018 in 63 Zentren in Nordamerika und Europa 352 Patienten (53% Männer; mittleres Alter 77 Jahre) eingeschlossen, davon 227 (64%) mit intrakranieller und 90 (26%) mit gastrointestinaler Blutung. Während der Rekrutierungsphase wurden die Einschlusskriterien durch „Protocol Amendments“ mehrfach modifiziert; so durften z.B. von Juli 2016 bis August 2017 nur Patienten mit intrakraniellen Blutungen eingeschlossen werden, um deren Anteil an der Studienpopulation zu erhöhen. Die häufigste Indikation für die Antikoagulation war VHF (80%). Andexanet wurde im Mittel 4,8 h nach Erstpräsentation in der Notfallabteilung verabreicht. Nur 254 Patienten (72%) erfüllten die Kriterien für die Analyse der Wirksamkeit (s.o.). Bei ihnen wurde nach Gabe von Andexanet eine signifikante Abnahme der Anti-Faktor-Xa-Aktivität gemessen (s. Tab. 1). Bei 5 Patienten erfolgte (ohne Angabe von Gründen) keine Evaluierung der Hämostase; von den verbliebenen 249 Patienten erreichten 202 (82%) eine “sehr gute“ (84%) oder „gute“ (16%) Stillung der Blutung nach 12 h. Innerhalb von 30 Tagen starben 49 Patienten (14%), davon zwei Drittel aus kardiovaskulären Ursachen. Thrombotische Ereignisse traten bei 34 Patienten auf (10%; Schlaganfall 14, Venenthrombose 13, Myokardinfarkt 7 und Lungenembolie 5 Patienten). Die Reduktion der Anti-Faktor-Xa-Aktivität war nicht prädiktiv für den hämostatischen Effekt in der gesamten Studienpopulation und nur mäßig prädiktiv in der Subgruppe mit intrakranieller Blutung. Bei keinem Patienten wurde eine Antikörperbildung gegen Andexanet nachgewiesen.
Schlussfolgerung und Diskussion: Die Autoren schließen aus ihren Ergebnissen, dass Notfallmedizinern in der Behandlung von Patienten mit lebensbedrohlichen Blutungen mit Andexanet ein wirksames und sicheres Antidot gegen direkte Faktor-Xa-Inhibitoren zur Verfügung steht. Die fehlende Korrelation des reinen Surrogatparameters „Anti-Faktor-Xa-Aktivität“ mit dem klinischen Endpunkt „Stillung der Blutung“ erklären die Autoren mit möglichen Störfaktoren (Confounders), wie z.B. nicht definierten Blutungsquellen (arteriell vs. venös) oder gestörter Thrombozytenfunktion. Gleichzeitig räumen sie aber ein, dass die Messung der Anti-Faktor-Xa-Aktivität nicht geeignet erscheint, einen klinischen Erfolg vorauszusagen.
Der größte Mangel der ANNEXA-4-Studie ist das Fehlen einer Kontrollgruppe. Nach Angaben der Autoren seien zum Zeitpunkt der Studienplanung „die logistischen und ethischen Herausforderungen“ für eine Plazebokontrolle zu groß und die Substitution von Gerinnungsfaktoren mit Prothrombinkomplex in dieser Indikation nicht zugelassen gewesen. Sie vergleichen allerdings in ihrer Diskussion ausführlich die Daten zur Wirksamkeit und Sicherheit von Andexanet in ANNEXA-4 mit denen von Prothrombinkomplex in publizierten Beobachtungsstudien mit ähnlichen (aber deutlich kleineren) Patientenpopulationen (6, 7). Soweit zu beurteilen, sind die Hämostaseraten in etwa gleich (um 70%); ein Vergleich von Letalität und Thromboseraten erscheint in Anbetracht der wenigen Patienten und heterogener Begleitumstände – z.B. Wiederbeginn der D/OAK – nicht sinnvoll. Letztlich ist ohne Kontrollgruppe weder eine aussagekräftige Beurteilung der Wirksamkeit noch der Sicherheit möglich.
Die Substitution mit Prothrombinkomplex ist zwar „off label“, wird mittlerweile aber in vielen Expertenstatements als Therapiemaßnahme bei bedrohlichen Blutungen unter DOAK empfohlen (z.B. 8, 9), ebenfalls ohne zugrundeliegende randomisierte kontrollierte Studien, und ist in der klinischen Praxis weit verbreitet. Diese Behandlung wäre als Kontrolltherapie naheliegend, ethisch durchaus vertretbar und daher für alle Studien zu DOAK-Antidoten zu fordern. Auch bleibt unklar, ob überhaupt ein Vorteil der DOAK-Antidote gegenüber alleinigen Standardmaßnahmen, z.B. DOAK-Pause, konservative/chirurgische Blutstillung, Transfusion, gegeben ist. Dies lässt sich aber wohl tatsächlich aus ethischen Gründen nicht mittels einer prospektiven Studie klären.
Der Andexanet-Hersteller kündigt die Durchführung einer randomisierten Phase-IV-Studie nach der Zulassung an. Sie soll Patienten mit intrakraniellen Blutungen einschließen, wie von der FDA im Rahmen des „Accelerated Approval Program“ als Auflage vorgeschrieben wurde. Nach den bekanntgegebenen Details (10) dürfte aber auch diese Studie keine standardisierte Vergleichstherapie beinhalten („Single group assignment“). Die Annahme liegt nahe, dass der Hersteller den Status quo möglichst lange aufrechterhalten möchte mit dem Ziel, den sehr teuren Wirkstoff (Einzelgabe Niedrigdosis: 29.000 US$, Hochdosis 58.000 US$; 11) bis zum Vorliegen (potenziell ungünstiger) klinischer Ergebnisse maximal zu vermarkten. Davon profitieren letztlich auch die DOAK-Hersteller, da mit der Verfügbarkeit spezifischer Antidots ein wesentliches Argument gegen die direkten Faktor-Xa-Inhibitoren wegfällt und diese als „noch sicherer“ verkauft werden können.
Als häufige Nebenwirkung von Andexanet (≥ 5%) werden in der US-Fachinformation Pneumonien und Harnwegsinfekte angeführt. Außerdem verfügte die FDA als eine Bedingung für die beschleunigte Zulassung eine „Boxed Warning“ aufgrund der in ANNEXA-4 berichteten kardiovaskulären Ereignisse.
EU-Marktzulassung: Der Ausschuss für Humanarzneimittel (Committee for Medicinal Products for Human Use = CHMP) der Europäischen Arzneimittel-Agentur (EMA) hat im Februar 2019 der Europäischen Kommission empfohlen, eine bedingte Zulassung („conditional marketing authorization“) für Andexanet auszusprechen (12). Mit dieser ist in den kommenden Monaten zu rechnen. Sie ist – analog zur beschleunigten Zulassung der FDA – an die Durchführung von weiteren Studien nach der Zulassung geknüpft und wird jährlich überprüft.
Fazit: Mit Andexanet wurde in den USA ein spezifisches Antidot gegen Faktor-Xa-Inhibitoren zugelassen. Eine baldige EU-Zulassung wird erwartet. Ähnlich wie beim einzigen bisher in der EU zugelassenen spezifischen DOAK-Antidot Idarucizumab gibt es zu Andexanet lediglich Daten aus nicht randomisierten Kohortenstudien ohne Kontrollgruppen (und mit beträchtlichen Veränderungen der Einschlusskriterien und Studienendpunkte im Verlauf von ANNEXA-4). Eine umfassende klinische Bewertung dieses neuen und teuren Antidots (z.B. vs. Prothrombinkomplex, aber auch vs. Standardtherapie) ist nicht möglich. Im Zusammenhang mit dem Akut-Management von Blutungskomplikationen unter DOAK bleiben weiterhin viele Fragen offen.
Literatur
- AMB 2019, 53, 17. Link zur Quelle
- January, C.T., et al.: J. Am. Coll. Cardiol. 2019. Link zur Quelle
- AMB 2017, 51, 92. Link zur Quelle
- Siegal, D.M., et al. (ANNEXA-A und ANNEXA-R = A phase 3 randomized, double-blind, placebo-controlled study in older subjects to assess safety and the reversal of apixaban anticoagulation with intravenously administered andexanet alfa): N. Engl. J. Med. 2015, 373, 2413. Link zur Quelle
- Conolly, S.J., et al. (ANNEXA-4 = Prospective, open-label study of andexanet alfa in patients receiving a factor Xa inhibitor who have acute major bleeding): N. Engl. J. Med. 2019, Feb 7. Link zur Quelle
- Majeed, A., et al.: Blood 2017, 130, 1706. Link zur Quelle
- Schulman, S., et al.: Thromb. Hemost. 2018, 118, 842. Link zur Quelle
- Maegele, M., et al.: Dtsch. Arztebl. Int. 2016, 113, 575. Link zur Quelle
- https://www.uptodate.com/contents/ management-of-bleeding-in- patients-receiving-direct-oral- anticoagulants Link zur Quelle
- ClinicalTrials.gov Identifier: NCT03661528
- https://www.uptodate.com/contents/ andexanet-alfa-drug-information Link zur Quelle
- https://www.ema.europa.eu/en/ news/first-antidote-reversal-anticoagulation -factor-xa-inhibitors- apixaban-rivaroxaban Link zur Quelle